Long QT (QTc) interval, long QT syndrome (LQTS) & torsades de pointes
The QT interval, long QT syndrome (LQTS) & ventricular arrhythmias (torsade de pointes) due to prolonged QT interval
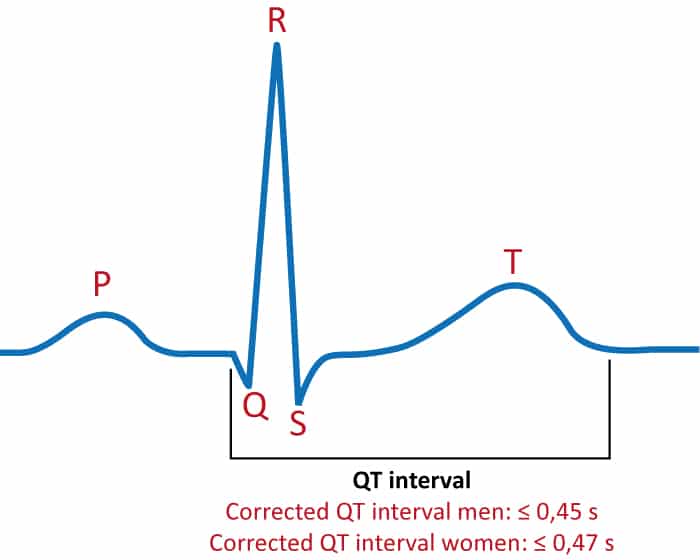
The QT interval is the time interval from the beginning of the QRS complex to the end of the T-wave. This interval represents the total time taken to depolarize and repolarize the ventricles (Figure 1). The length of the QT interval correlates strongly with the risk of potentially life-threatening ventricular arrhythmias. Therefore, the QT interval must always be assessed when interpreting the ECG. Long QT syndrome (LQTS) is manifest when a long QT interval induces ventricular arrhythmias.
The QT interval is inversely related to heart rate. As the heart rate increases, the QT interval decreases and vice versa. The physiological purpose of this phenomenon is to allow for faster cardiac cycles during tachycardia (e.g during physical exertion). Therefore, to judge whether the QT interval is normal or not, one must adjust for the current heart rate. This is done by adjusting the QT interval for the heart rate, and the resulting QT interval is referred to as corrected QT interval, or simply QTc interval. The primary hazard lies in long QTc intervals because they induce a very unstable polymorphic ventricular tachycardia referred to as torsade de pointes. Abnormally short QTc interval is also arrhythmogenic but it is a very rare condition.
Several formulas have been suggested to calculate corrected QT intervals. Some of these formulas follow:
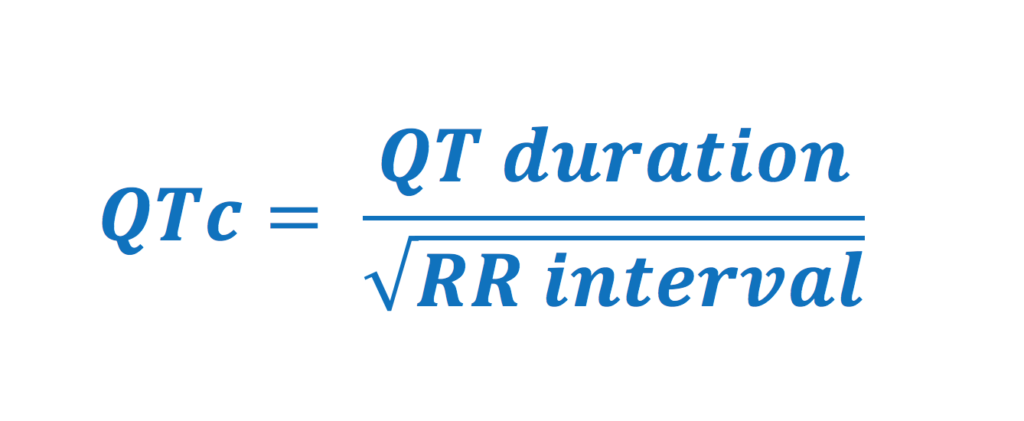
Bazett Formula: QTc = QT interval / √(RR interval)
Fridericia Formula: QTc = QT interval / (RR interval)1/3
Framingham Formula: QTc = QT interval + 154 x (1 – RR interval)
Hodges Formula: QTc = QT interval + 1.75 x [(60 / RR interval) − 60]
RR interval = 60 / HR
Calculate corrected QTc interval
Bazett’s formula is the most commonly used formula. However, all above-listed formulas were developed many decades ago and they have multiple drawbacks. For example, Bazett’s formula is only suitable in adults with a heart rate between 60 and 90 beats per minute. Bazett’s formula over-adjusts at higher heart rates and under-adjusts at lower heart rates.
| 1 to 15 years, male and female | Adult, male | Adult, female | |
|---|---|---|---|
| Normal | <440 ms | <430 ms | <450 ms |
| Upper limit | 440–460 ms | 430–450 ms | 450–470 ms |
| Prolonged | >460 ms | >450 ms | >470 ms |
It is recommended that the automatic (machine) calculation of the corrected QT interval be used. Such QTc intervals are derived by all modern ECG machines and the formulas used are more precise than those listed above. It is recommended that whenever the QTc interval is prolonged, it should be verified manually.
How to measure the QT interval
Expert opinion suggests that QT intervals should be measured as follows (Anderson et al):
- The QT interval should be measured manually, from the beginning of the QRS complex to the end of the T wave.
- The QT interval should be measured in 3 to 5 consecutive beats in leads II, V5 and V6. Leads without U waves are preferred.
- Calculate the mean QT interval for each lead and use the longest QT interval obtained.
- Large U waves that are fused with the T wave should be included in the measurement. This may result in an overestimation of the QT interval.
- If the patient uses QT-prolonging drugs, the QT interval should be measured during peak plasma concentrations of that drug.
- The QT interval should be adjusted for heart rate.

Long QT interval causes long QT syndrome
An abnormally prolonged QTc interval is referred to as long QT interval. The upper reference limit for QTc interval is 460 ms in males and 470 ms in females. QTc intervals exceeding these limits may cause torsade de pointes. If this occurs, i.e if a person with a long QT interval experiences such ventricular arrhythmias, the condition is referred to as long QT syndrome (LQTS).
Causes of long QT interval
A long QT interval is either congenital (genetic) or acquired.
Congenital long QT syndrome is caused by mutations in cardiac ion channels. More than 10 types of congenital prolongation of the QT interval have been discovered. Congenital QT prolongation is a very serious condition with high mortality. Among untreated patients who have experienced one episode of syncope, 20% die within 1 year. Fortunately, this mortality figure may be reduced to 1% over 15 years of follow-up with the use of evidence-based treatments. Three types of LQTS (LQT1, LQT2 and LQT3) represent roughly 90% of all cases of congenital LQTS. It is estimated that the prevalence of congenital QT prolongation is 1 per 2000 individuals in the population (prevalence figures from Italy). Importantly, individuals with congenital QT prolongation frequently report occurrences of unexplained syncope or cardiac arrest in the family. Such hereditary information is a strong predictor of sudden cardiac death.
Acquired long QT syndrome is caused by medications (amiodarone, sotalol, procainamide), hypokalemia, hypomagnesaemia and pronounced bradycardia. Because each of these factors (medications, electrolyte disorders and bradycardia) are all common, but only appear to cause QT prolongation in some individuals, it is thought that there has to be an underlying genetic susceptibility to develop acquired long QT syndrome.
The risk of developing torsade de pointes (polymorphic ventricular tachycardia) is evident in both congenital and acquired QT prolongation. The longer the QT interval the greater the risk of developing torsade de pointes. In general, torsade de pointes develops at QTc intervals greater than 490 milliseconds.
Torsade de pointes is usually induced by a premature ventricular beat occurring early in the cardiac cycle. The risk of torsade de pointes increases during bradycardia. Torsade de pointes causes syncope (or pre-syncope) but the arrhythmia is usually self-terminating (within 30 seconds). A minority of cases of torsade de pointes progress to ventricular fibrillation, which is fatal unless treatment is given promptly. Figure 2 shows torsade de pointed.
Log in to view image, video, quiz, text

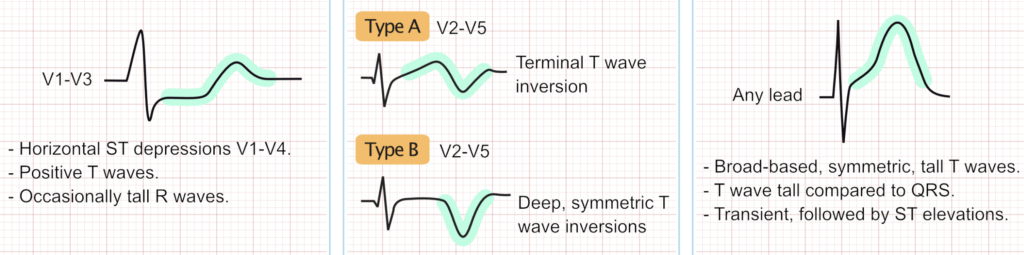
Besides the QT interval itself, the T-wave may provide valuable information regarding the type of long QT syndrome; particularly it may differentiate between type 1 LQTS, type 2 LQTS and type 3 LQTS. The T-wave should be assessed in the chest leads. Refer to Figure 3. Occasionally persons with LQTS display T-wave alternans, meaning that the amplitude or direction of the T-wave alternates from one beat to the next. T-wave alternans is an indicator of very high risk of torsade de pointes. Sinus pauses may also occur in congenital LQTS.
ECG criteria for torsade de pointes
- Prolonged QTc interval before the appearance of torsade de pointes.
- Twisting of the QRS complexes around the isoelectric baseline (polymorphic ventricular tachycardia).
Log in to view image, video, quiz, text
Log in to view image, video, quiz, text
Congenital long QT syndrome (LQTS)
At least 13 variants of congenital LQTS have been described. The mutations have autosomal inheritance with reduced penetrance. LQTS type 1, type 2 and type 3 (called LQT1, LQT2 and LQT3) represent 90% of all cases of long QT syndrome. LQT1 and LQT2 each represent roughly 40% of all cases.
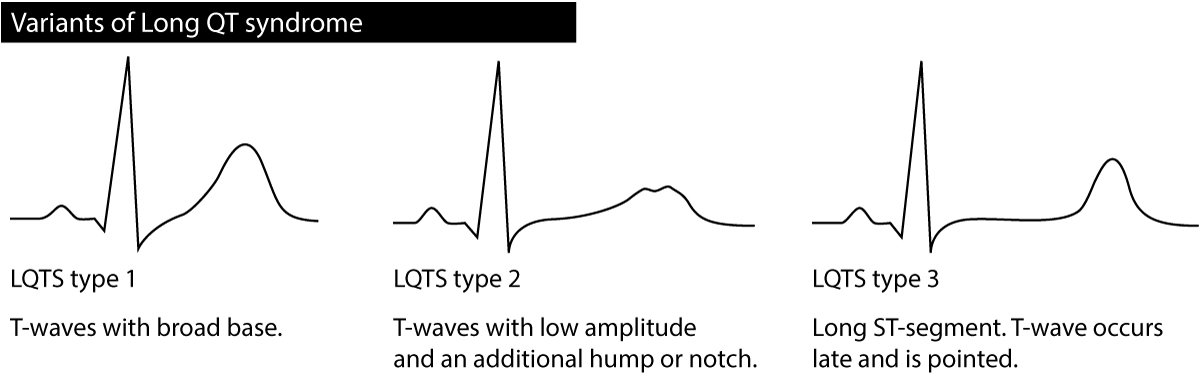
Long QT syndrome type 1 (LQT1) is caused by a mutation in the potassium channel KCNQ1 (loss of function mutation). The arrhythmias usually occur during physical activity (for some reason swimming appears to be highly arrhythmogenic) and other situations with high sympathetic activity. LQT1 is characterized by T-wave with a broad base (Figure 3). LQT1 is the most common type of congenital LQTS.
Long QT syndrome type 2 (LQT2) is caused by a mutation in the potassium channel KCNH2 (loss of function mutation). The arrhythmias typically occur at sudden surprises (sudden sounds, fear or other situations with abrupt and sudden stress), stress, physical activity or during sleep. The T-wave has low amplitude with an additional hump or notch (Figure 3). Women with LQT2 who are in the postpartum period have very high risk of developing torsade de pointes.
Long QT syndrome type 3 (LQT3): is caused by a mutation in the sodium channel SCN5A (leads to increased sodium flows). The risk of arrhythmia is highest during sleep. Bradycardia is also highly arrhytmogenic in these patients. The ST-segment is stretched out, the T-wave occurs late and is pointed (Figure 3).
Long QT syndrome type 4 (LQT4): is rare and represents 1% of all cases. The mutation occurs in the ANKB gene which produces a protein that anchors membrane proteins to the cytoskeleton. LQT3 can cause multiple arrhythmias, such as familial catecholamine ventricular tachycardia, atrial fibrillation, conduction defects, sinus node dysfunction and bradycardia.
The other variants of LQTS are extremely rare and usually part of more severe syndromes which engage multiple organ systems. These types are not discussed here.
Schwartz criteria for diagnosis of congenital LQTS
Schwartz criteria are used to diagnose congenital LQTS. These criteria are presented in Table 1.
Table 1. Long QT syndrome diagnostic criteria (Schwartz et al):
| ECG findings | CRITERIA | POINTS |
|---|---|---|
| QTc interval | ≥480 ms | 3 |
| 460–479 ms | 2 | |
| 450–459 ms (male) | 1 | |
| QTc during 4th minute of recovery from exercise stress test ≥480 ms | 1 | |
| Torsade de pointes | 2 | |
| T-wave alternans | 1 | |
| Low heart rate for age (resting heart rate below the 2nd percentile) | 0.5 | |
| Clinical History | ||
| Syncope with stress | 2 | |
| Syncope without stress | 1 | |
| Congenital deafness | 0.5 | |
| Family history | ||
| Family members with definite LQTS | 1 | |
| Unexplained sudden cardiac death below age 30 among immediate family members | 0.5 |
Evaluation of risk
- ≤1 point: low probability of LQTS.
- 1.5 – 3 points: intermediate probability of LQTS.
- ≥3.5 points: high probability.
Notes
- ECG findings are only valid in the absence of medications or disorders known to affect these electrocardiographic features.
- QTc is calculated by Bazett’s formula where QTc = QT/√RR.
- Only one of syncope and torsade de pointes can count.
- The same family member cannot be counted in both criteria under Family history.
Long QT syndrome induced by medications and drugs
Long QT syndrome caused by medications is much more common than congenital variants. Medications that may induce or aggravate long QT syndrome include adrenaline, certain antihistamines, erythromycine, trimetoprime, sulfa, pentamidine, kinidine, procainamide, disopyramide, sotalol, probukol, bepridil, difetilid, ibutilid, cisaprid, ketokonazol, itrakonazol, tricyclic antidepressants, fenotiazines, haloperidol, indapimid, certain antiviral drugs, etc (Table 2). The list of drugs that cause LQTS is very long and updated continuously. The complete list is provided by CredibleMeds (www.crediblemeds.com) which is supported by the FDA.
Table 2. Drugs causing or aggravating long QT syndrome.
| CLASS | DRUG | ASSOCIATION | RISK OF TORSADE DE POINTES | EFFECT | COMMENTS |
|---|---|---|---|---|---|
| Anesthetics | Enflurane | Probable | Drug-drug interactions lead to QT prolongation | ||
| Halothane | Probable | Nonspecific arrhythmias reported in package insert. | |||
| Isoflurane | Probable | ||||
| Antiarrhythmics | Amiodarone | Certain | High | QT interval prolongation, Torsade de pointes | i.v. affects QTc less than oral; proarrhythmia infrequent. |
| Adenosine | Proposed | ||||
| Disopyramide | Certain | QT interval prolongation, Torsade de pointes | Rate appears lower than that of quinidine | ||
| Dofetilide | Certain | High | QT interval prolongation, Torsade de pointes | Proarrhythmia 0.8%. | |
| Flecainide | Certain | High | QT interval prolongation, Torsade de pointes | Proarrhythmia “rare.” | |
| Ibutilide | Certain | High | QT interval prolongation, Torsade de pointes | Proarrhythmia 1.7%. | |
| Procainamide | Certain | High | QT interval prolongation, Torsade de pointes | Rate appears lower than that of quinidine. | |
| Propafenone | Certain | Moderate | QT interval prolongation, Torsade de pointes | Proarrhythmia “rare.” | |
| Quinidine | Certain | High | QT interval prolongation, Torsade de pointes | “Quinidine syncope” in 2–6% of patients. | |
| Sotalol | Certain | High | QT interval prolongation, Torsade de pointes | Proarrhythmia ~2%. | |
| Anticonvulsants | Felbamate | Proposed | Torsade de pointes, according to manufacturer. | ||
| Fosphenytoin | Proposed | QT interval prolongation according to manufacturer. | |||
| Antidepressants | Amitriptyline | Certain | Moderate | Nonspecific ECG changes reported in package insert. | |
| Citalopram | Probable | ||||
| Desipramine | Certain | QT interval prolongation | VF, sudden death reported by manufacturer. | ||
| Doxepin | Certain | Moderate | |||
| Fluoxetine | Probable | QT interval prolongation, Torsade de pointes | 1 in 10,000 ventricular arrhythmias reported by manufacturer. | ||
| Imipramine | Certain | Moderate | Nonspecific arrhythmias reported in package insert. | ||
| Maprotiline | Certain | ECG changes; QRS reported in package insert. | |||
| Nortriptyline | Certain | Nonspecific arrhythmias reported in package insert. | |||
| Paroxetine | Probable | Torsade de pointes | Lower risk than that of TCAs. | ||
| Sertraline | Probable | QT interval prolongation, Torsade de pointes | Lower risk than that of TCAs. | ||
| Venlafaxine | Proposed | QT interval prolongation | 1:1000 risk of arrhythmia reported in package insert. | ||
| Antihistamines | Astemizole | Certain | Moderate | Not applicable | |
| Clemastine | Proposed | ||||
| Diphenhydramine | Proposed | ||||
| Loratadine | Proposed | Unknown | Prolongation appears unlikely. | ||
| Terfenadine | Certain | Moderate | |||
| Antiinfectives | Clarithromycin | Probable | Moderate | QT interval prolongation, Torsade de pointes | |
| Erythromycin | Certain | Medium high | QT interval prolongation, Torsade de pointes | Known drug-drug interactions with other agents (e.g., terfenadine). | |
| Fluconazole | Probable | Risk may be higher with i.v. dosing. | |||
| Foscarnet | Proposed | QT interval prolongation | |||
| Ganciclovir | Proposed | ||||
| Gatifloxacin | Probable | QT interval prolongation | |||
| Grepafloxacin | Certain | ||||
| Halofantrine | Certain | Moderate | |||
| Ketoconazole | Probable | QT interval prolongation, Torsade de pointes | Known drug-drug interactions with other agents (e.g., cisapride). | ||
| Levofloxacin | Proposed | Torsade de pointes | Lower risk than that of similar agents. | ||
| Mefloquine | Proposed | QT with halofantrine. | |||
| Moxifloxacin | Probable | QT interval prolongation | Lower risk than that of similar agents. | ||
| Pentamidine | Certain | Moderate | QT interval prolongation, Torsade de pointes | ||
| Quinine | Probable | Moderate | |||
| Sparfloxacin | Certain | ||||
| Trimethoprim-sulfamethoxazole | Proposed | Low | |||
| Antipsychotics | Chlorpromazine | Probable | Nonspecific ECG changes and sudden death reported by manufacturer. | ||
| Clozapine | Proposed | ||||
| Haloperidol | Certain | Moderate | QT interval prolongation, Torsade de pointes | ||
| Mesoridazine | Certain | QT interval prolongation, Torsade de pointes | |||
| Pimozide | Certain | QT interval prolongation, Torsade de pointes | Drug-drug interactions also lead to QT prolongation. | ||
| Quetiapine | Proposed | QT interval prolongation | |||
| Risperidone | Proposed | QT interval prolongation | Sudden death reported by manufacturer. | ||
| Sertindole | Proposed | Moderate | |||
| Thioridazine | Certain | Moderate | QT interval prolongation, Torsade de pointes | ||
| Ziprasidone | Certain | QT interval prolongation | |||
| Cancer drugs | Arsenictrioxide | Certain | QT interval prolongation, Torsade de pointes | ||
| Tamoxifen | Probable | QT interval prolongation | Overdose situations. | ||
| Cardiovascular agents | Bepridil | Certain | Moderate | QT interval prolongation, Torsade de pointes | QTc increases by ~8%. |
| Indapamide | Proposed | QT interval prolongation | |||
| Isradipine | Proposed | QT interval prolongation according to manufacturer. | QTc increases by ~3%. | ||
| Mibefradil | Proposed | ||||
| Moexipril hydrochlorothiazide | Proposed | QT interval prolongation according to manufacturer. | |||
| Nicardipine | Proposed | QT interval prolongation according to manufacturer. | |||
| Probucol | Certain | ||||
| Gatrointestinal agents | Cisapride | Certain | Moderate | QT interval prolongation, Torsade de pointes | |
| Octreotide | Proposed | QT interval prolongation | |||
| Droperidol | Certain | Certain | QT interval prolongation, Torsade de pointes | ||
| Dolasetron | Proposed | QT interval prolongation according to manufacturer. | |||
| Migraine agents | Naratriptan | Probable | QT interval prolongation | ||
| Sumatriptan | Probable | QT interval prolongation | |||
| Rizatriptan | Probable | 1:1000 risk of arrhythmia. | |||
| Zolmitriptan | Probable | QT interval prolongation | |||
| Miscellaneous agents | Amantadine | Low | |||
| Epinephrine | Proposed | ||||
| Levomethadyl | Probable | QT interval prolongation, Torsade de pointes | |||
| Methadone | Probable | Syncope according to manufacturer. | |||
| Salmeterol | Proposed | QT interval prolongation according to manufacturer. | |||
| Tacrolimus | Proposed | ||||
| Tizanidine | Probable | QT interval prolongation | 1:1000 risk of arrhythmia. |
Management of long QT syndrome (LQTS)
Torsade de pointes with hemodynamic compromise
Torsade de pointes causing syncope is treated with defibrillation. Start with 150 J (biphasic shock) and increase by 50 J for each shock. Ventricular fibrillation and cardiac arrest are treated with conventional resuscitation.
Hemodynamically stable torsade de pointes
Treatment of torsade de pointes is similar in congenital and acquired LQTS. Torsade de pointes is paroxysmal, which means that the arrhythmia occurs intermittently and self-terminates. It tends to recur, even after successful defibrillation. There is always risk of ventricular fibrillation which is why a defibrillator must be close at hand and readiness to perform resuscitation is necessary.
Treatment algorithm
- All medications/drugs that may cause or aggravate the arrhythmia must immediately be stopped.
- Magnesium infusion (regardless of blood magnesium levels): 1 gram of magnesium is administered intravenously for 60 seconds. This may be repeated after 5–10 minutes. If a continuous infusion is necessary, the dose is 5–10 mg/min.
- Potassium infusion: only necessary if the patient has hypokalemia.
- Bradycardia must be corrected: bradycardia may induce and aggravate torsade de pointes. To correct bradycardia, the following options are at hand:
- atropine i.v 1–2 ml 0.5 mg/ml.
- isoprenaline (isoproterenol) 0.01 μg/kg/min, which is titrated up until bradycardia resolves. Note that isoprenaline must be administered carefully because it activates beta-adrenergic receptors which is why it may aggravate the arrhythmia. In congenital LQTS isoprenaline is contraindicated because the risk of ventricular fibrillation is high. Therefore, isoprenaline may only be used in acquired LQTS and temporarily until a pacemaker can be established.
- temporary transcutaneous/transvenous pacemaker. The pacemaker electrode should be placed in the atria and the rate should be set to 90 beats per minute. The rate may be increased gradually until the arrhythmia resolves.
The rationale behind atropine, isoprenaline and pacemaker therapy is simple: these three interventions all increase the heart rate, which decreases the QTc interval and thus terminates the torsade de pointes.
Long-term treatment for acquired long QT syndrome
No treatment is necessary after the removal of the medications causing the syndrome.
Long-term treatment for congenital long QT syndrome
Beta-blockers are very effective in congenital LQTS. Mortality is reduced dramatically if the right drug and right dose is given. Propranolol (usually sufficient with 3 mg/kg/day) and nadolol (usually sufficient with 1 mg/kg/day) are the most effective drugs. Metoprolol has a proven effect but is less effective than propranolol and nadolol. There are no studies available on atenolol, which therefore cannot be recommended. Patients with pronounced bradycardia should not be given beta-blockers due to the risk of provoking torsade de pointes.
Artifical pacemaker may be necessary if the maximal dose of beta-blockers is insufficient. If pacemaker is also insufficient, sympathectomy may be considered. Sympathectomy means that sympathetic nerve ganglia (thoracic) are removed surgically, which leads to the elimination of adrenergic stimulation to the heart. This is an effective method but requires surgery.
ICD (Intracardial Cardiac Defibrillator) is used in the following cases:
- Patients who have experienced cardiac arrest.
- Patients who have experienced syncope despite optimal treatment (maximal dose beta blocker, pacemaker and possibly sympathectomy).
- If the anamnesis is very worrying and the QTc interval is >550 ms. T-wave alternans and sinus pause corroborates this further.
Short QT syndrome (SQTS)
Short QT syndrome is extremely rare but may cause polymorphic ventricular tachycardia. It is defined as QTc interval <0.35 s. Note that hypercalcemia and digoxin which may also shorten the QTc interval.