The course from sudden cardiac arrest (SCA) to death follows a pathophysiological process that affects the effectiveness of the interventions. Numerous studies demonstrate that the available interventions are time-sensitive, and to maximize the likelihood of restoring spontaneous circulation, interventions should be adapted to the current phase. However, current guidelines do not consider this fact (beyond the recommendation to defibrillate immediately if cardiac arrest is witnessed and the rhythm is shockable). This is explained by the fact that there are no means of determining what phase the patient is in. The three phases following a sudden cardiac arrest were described by Weisfeldt and Becker (2002). Drawing on physiology, observational and experimental studies, they described these phases, which require different treatment strategies to maximize survival (Table 1).
| Phase | Period | Optimal treatment |
|---|---|---|
| Electrical phase | 0 to 4 minutes | Defibrillation |
| Circulatory phase | 4 to 10 minutes | Compressions and ventilation, followed by defibrillation |
| Metabolic phase | >10 minutes | Unknown |
The existence of these stages is supported by a wide range of experimental and clinical studies. The electrical phase and circulatory phase are explained by cellular changes in the myocardium. The last phase is multifactorial and caused by global ischemia, acidosis, inflammation, hypoxia, and reperfusion injury.
Myocardial oxygen demand
Myocardium can only function under aerobic conditions (oxygen is required for ATP synthesis [ATP is the primary energy source in all cells]). The heart consumes approximately 30 kilograms of ATP per day (Neubauer et al), which is about 100 times its weight. This is possible because mitochondria occupy nearly 30% of the myocardial cell volume, which enables the generation of such large amounts of ATP (Page et al). The mitochondrial density allows for complete replenishment of ATP within 10 seconds (Fell et al, Houtkooper et al). The majority of ATP produced is utilized to fuel the contractile machinery and maintain ion channel function.
Neumar et al studied how the ATP concentration diminishes during ventricular fibrillation (VF). This was done by taking serial myocardial biopsies after inducing VF. As evident in Figure 1, ATP concentrations decline rapidly. After 5 minutes of VF, approximately 50% of the ATP remained.

As ATP concentration declines, critical cell functions cease. Membrane potential, action potential, excitability, and contractile function are all compromised by ATP depletion. Myocardial necrosis (infarction) commences within 20 minutes of circulatory collapse.
The electrical phase: Defibrillate
It is likely that the majority of cardiac arrests are triggered by ventricular tachycardia (VT) and ventricular fibrillation (VF). Defibrillation is extremely effective during the first few minutes of these arrhythmias. This is evident from randomized trials and observational studies examining the efficacy of ICD (implantable cardioverter defibrillator). An ICD is programmed to detect VF/VT and defibrillate within 10—20 seconds. Studies show that ICDs succeed in terminating VF/VT in 98% of cases (Zipes et al, Volosin et al). Similarly, in individuals who are hospitalized when a cardiac arrest occurs, VF/VT can be detected and defibrillated promptly. Survival is around 70% when defibrillation is performed within 3 minutes of the collapse (Hessulf et al). In out-of-hospital cardiac arrest (OHCA), the delay from collapse to defibrillation is substantially longer. The median time to arrival of the EMS is 10 to 15 minutes in Europe and North America, resulting in a survival rate of approximately 30% if the rhythm is shockable upon the arrival of EMS (Rawshani et al).
| Delay from VT/VF onset to defibrillation | Evidence | Success rate of defibrillation |
|---|---|---|
| <30 seconds | ICD trials | 98% |
| 3 minutes | Observational studies | 70% |
| <10–15 minutes | Observational studies | 20–30% |
With each passing minute, the likelihood of successful defibrillation decreases and the risk of VF degenerating into asystole increases. Figure 2 shows how the survival rate drops from 42% to 29% in attempts to defibrillate after 5 minutes when CPR is started after 1 and 5 minutes respectively (out-of-hospital cardiac arrest with VF; Valenzula et al).
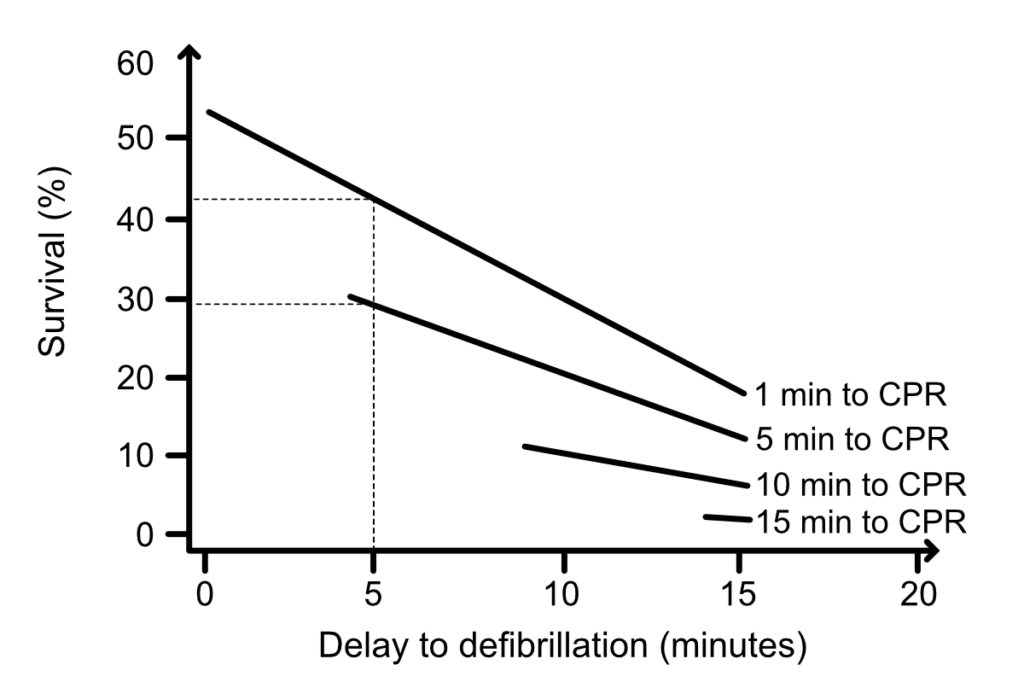
Undoubtedly, the effect of defibrillation is very high during the first few minutes of VF. During this period (0–4 minutes), compressions are presumably of little or no utility (Niemann et al). Thus, an electric shock is the most appropriate initial intervention during the electrical phase.
The circulatory phase: Compress and ventilate
In 1943, Gurvich et al demonstrated that the probability of successfully defibrillating VF after several minutes increased if defibrillation was preceded by chest compressions. As can be seen in Figure 2, defibrillation is less effective as the delay in initiating compressions increases. The circulatory phase starts approximately after 4 minutes and during this phase, defibrillation is much less effective. Compressions are critical for myocardial perfusion, which is required to render the myocardium amenable to defibrillation. The likelihood of successfully defibrillating VF increases if compressions precede defibrillation during this phase. This has been demonstrated in several animal studies:
- Yakaitis et al showed that defibrillation was the most critical first intervention if performed within 3 minutes of collapse. After 5 minutes of VF, defibrillation succeeded in only 30% of cases, compared to 70% when compressions preceded the defibrillation.
- Niemann et al showed that if VF continued for 7 minutes, defibrillation was 3 times more effective if preceded by compressions.
- Menegazzi et al showed that if VF continued for 8 minutes, defibrillation was 3 times more effective if preceded by compressions and antiarrhythmics.
- Garcia et al showed that defibrillation was ineffective if VF continued for 6 minutes.
Thus, several experimental animal studies show that the longer VF is present, the lower the likelihood of successful defibrillation. This certainly holds true in humans also. Fortunately, it can be countered by performing compressions and ventilation. The compressions are critical in generating the coronary perfusion pressure (CPP) required in order to normalize the electrical potential of myocardial cells. Indeed, after a few minutes, the myocardium becomes so sensitive to interruptions in perfusion that the time it takes for the defibrillator to charge increases mortality. Yu et al demonstrated this by intentionally delaying defibrillation by 3, 10, 15, or 20 seconds (pause after compressions); all defibrillations succeeded if the interruption was 3 seconds long, but none were successful if the interruption was 15 seconds or longer. After an interruption in compressions, it takes about 15 seconds of compressions to rebuild the coronary perfusion pressure.
When Cobb et al examined the effect of performing compressions during 90 seconds before defibrillation in out-of-hospital cardiac arrest, a 42% higher probability of survival was noted (cases presenting with VF and in whom EMS response times were 4 minutes or longer). A Norwegian study involving 200 out-of-hospital cardiac arrests similarly showed that 3 minutes of CPR before defibrillation increased the likelihood of ROSC by 2 times (when EMS response time was longer than 5 minutes; Wik et al).
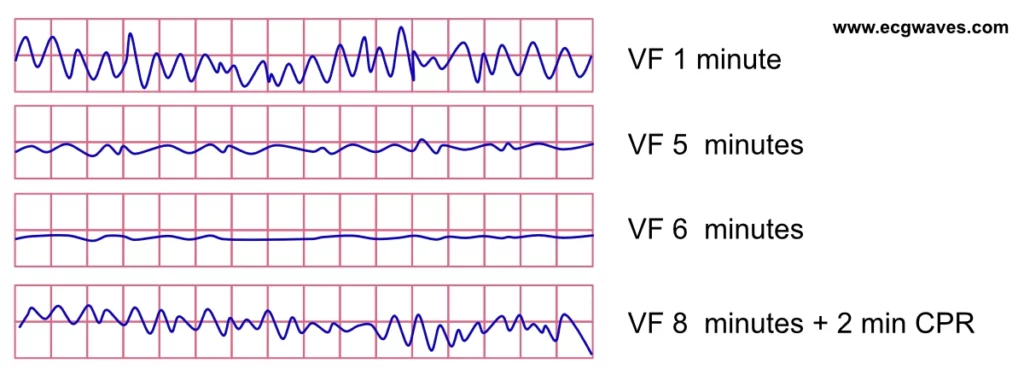
The ECG reflects the pathophysiological process that renders defibrillation ineffective after several minutes of ventricular fibrillation. In parallel with the reduction of ATP concentration in the myocardium, the amplitude of the VF waveform decreases. The ECG demonstrates a gradual transition from coarse VF to fine VF. Fine VF ultimately transitions to asystole and death.
The metabolic phase
Most deaths occur during the metabolic phase, which begins approximately after 10 minutes. However, biological death (brain death) occurs after 6–8 minutes of total circulatory arrest. Individuals who survive cardiac arrest with longer durations have special circumstances that extend the time to biological death (e.g. periods of ventricular tachycardia that generate cardiac output, young age, accidental hypothermia, etc.). If cardiopulmonary resuscitation is started during the metabolic phase, compressions and defibrillation are usually insufficient to save the individual. It is unclear which treatment is optimal during this phase.
The metabolic phase is characterized by extensive cerebral cell death and impending myocardial necrosis. Myocardial ATP levels are so low that electrical activity has ceased (asystole occurs). The remaining tissues have developed ischemia with subsequent local and global acidosis. Effective compressions can then result in reperfusion damage that exacerbates tissue damage. There are data suggesting that reperfusion in the myocardium accelerates myocardial necrosis (Vanden Hoek et al). In addition, there are pronounced electrolyte disturbances that complicate the possibility of maintaining ROSC.
During the metabolic phase, brain damage and myocardial infarction develop both by ischemia (which causes cell death by cessation of critical cell functions) and toxic effects of reperfusion (induced by CPR). Reperfusion damage occurs when oxygen transmission to the tissue results in the production of reactive oxygen (ROS) compounds. Reperfusion damage only occurs if it lasts too long for oxygen delivery; there is an unknown time limit when oxygen delivery leads to tissue damage.
References
Kaustubha D. Patil, Henry R. Halperin, Lance B. Becker. Cardiac Arrest Resuscitation and Reperfusion. Circulation Research (2015).
Neubauer S. The failing heart—An engine out of fuel. New England Journal of Medicine. 2007;356:1140-1151
Fell DA, Sauro HM. Metabolic control analysis. The effects of high enzyme concentrations. European Journal of Biochemistry. 1990;192:183-187
Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of nad: An old metabolite controlling new metabolic signaling pathways. Endocrine Reviews. 2010;31:194-223
Gurvich NL, Yuniev GS. Restoration of heart rhythm during fibrillation by a condenser discharge. Am Rev Sov Med 1947;4:252–6.
Vanden Hoek TL, Shao Z, Li C, Zak R, Schumacker PT, Becker LB. Reperfusion injury in cardiac myocytes after simulated ischemia. Am J Physiol. 1996;270: H1334-H1341. 30.
Vanden Hoek TL, Qin Y, Wojcik K, et al. Reperfusion, not simulated ischemia, initiates intrinsic apoptosis injury in chick cardiomyocytes. Am J Physiol Heart Circ Physiol.
Yakaitis RW, Ewy GA, Otto CW, Taren DL, Moon TE. Influence of time and therapy on ventricular defibrillation in dogs. Crit Care Med. 1980;8:157-163.
Menegazzi JJ, Davis EA, Yealy DM, et al. An experimental algorithm versus standard advanced cardiac life support in a swine model of out-of-hospital cardiac arrest. Ann Emerg Med. 1993;22:235-239. 23.
Menegazzi JJ, Seaberg DC, Yealy DM, Davis EA, MacLeod BA. Combination pharmacotherapy with delayed countershock vs standard advanced cardiac life support after prolonged ventricular fibrillation. Prehosp Emerg Care. 2000;4:31-37.
Niemann JT, Cairns CB, Sharma J, Lewis RJ. Treatment of prolonged ventricular fibrillation. Circulation. 1992;85:281-287. 20.
Niemann JT, Cruz B, Garner D, Lewis RJ. Immediate countershock versus cardiopulmonary resuscitation before countershock in a 5-minute swine model of ventricular fibrillation arrest. Ann Emerg Med. 2000;36:543-546.
Garcia LA, Allan JJ, Kerber RE. Interactions between CPR and defibrillation waveforms. Resuscitation. 2000;47:301-305.
Page E, McCallister LP. Quantitative electron microscopic description of heart muscle cells. Application to normal, hypertrophied and thyroxin-stimulated hearts. Am J Cardiol. 1973;31:172–181.