ST-Elevation Myocardial Infarction (STEMI): Pathophysiology, Diagnosis, and Management
Coronary Atherosclerosis
Coronary atherosclerosis is a chronic, progressive, and highly complex lipid-driven inflammatory disease of the arterial intima that serves as the fundamental pathophysiological substrate for acute coronary syndromes (ACS), including ST-Elevation Myocardial Infarction (STEMI) [1]. Historically viewed as a simple plumbing problem characterized by the gradual accumulation of inert cholesterol debris, modern vascular biology has redefined atherosclerosis as a dynamic, immune-mediated fibroproliferative response to endothelial injury [2]. The process initiates with endothelial dysfunction, a state provoked by systemic risk factors such as hypertension, hyperglycemia, smoking, and turbulent hemodynamic shear stress. This dysfunction compromises the endothelial barrier, upregulating the expression of vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), and increasing the permeability of the endothelium to circulating apolipoprotein B (ApoB)-containing lipoproteins, predominantly low-density lipoprotein cholesterol (LDL-C) [3].
Once trapped within the subendothelial space, LDL-C particles undergo oxidative modification by reactive oxygen species (ROS) and enzymes such as myeloperoxidase and lipoxygenase. Oxidized LDL (oxLDL) is highly immunogenic and acts as a potent chemoattractant. Circulating monocytes bind to the upregulated adhesion molecules, transmigrate into the intima via diapedesis, and differentiate into tissue macrophages under the influence of macrophage colony-stimulating factor (M-CSF) [4]. These macrophages express scavenger receptors (e.g., SR-A, CD36) that, unlike the classic LDL receptor, are not downregulated by intracellular cholesterol accumulation. Consequently, macrophages indiscriminately phagocytose oxLDL until they become engorged with lipid droplets, transforming into characteristic foam cells. The accumulation of these foam cells forms the earliest macroscopically visible lesion of atherosclerosis: the fatty streak [5].
Over decades, the lesion progresses from a simple fatty streak to a complex fibroatheroma. Smooth muscle cells (SMCs) migrate from the tunica media into the intima, driven by platelet-derived growth factor (PDGF) and other cytokines. Within the intima, SMCs undergo a phenotypic switch from a contractile to a synthetic state, producing abundant extracellular matrix (ECM) components, primarily interstitial collagen types I and III, elastin, and proteoglycans [6]. This matrix forms a fibrous cap that sequesters the highly thrombogenic lipid core from the flowing blood. While atherosclerosis progresses indolently and non-linearly, often remaining asymptomatic for decades, it sets the anatomical and biochemical stage for sudden, life-threatening acute coronary syndromes when the structural stability of the plaque is acutely compromised [7]. Sub-group analyses from longitudinal angiographic registries demonstrate that the severity of baseline stenosis does not reliably predict the site of future STEMI; rather, it is the biological composition and inflammatory activity of the plaque that dictate clinical risk [8].
Plaques
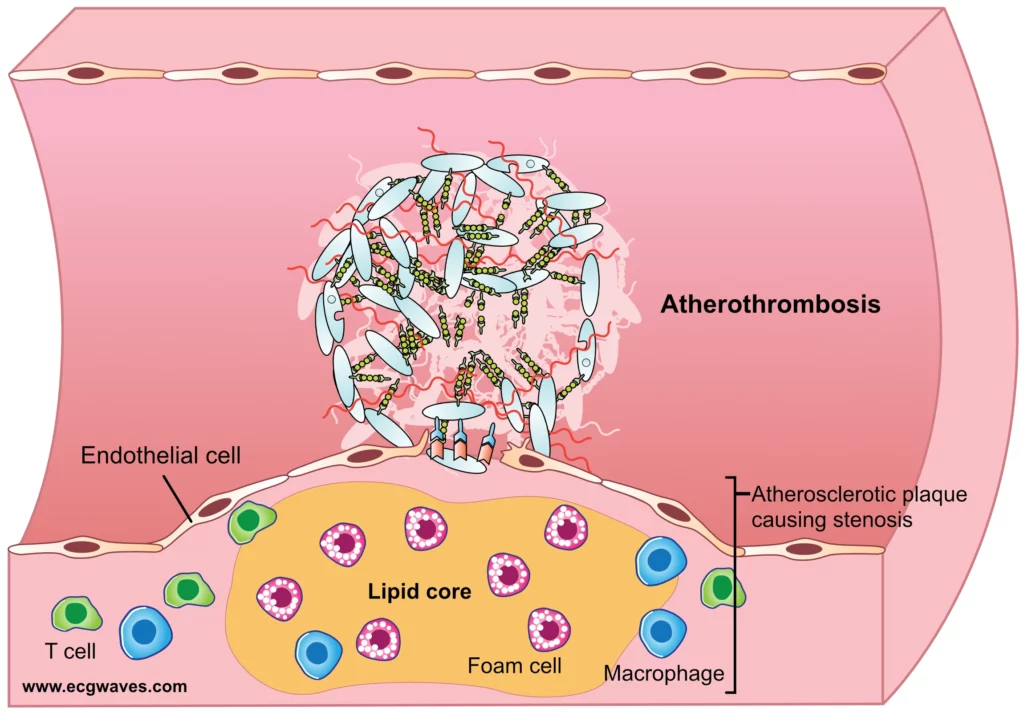
The architecture and composition of the atherosclerotic plaque are the primary determinants of its vulnerability to disruption. Advanced atherosclerotic plaques consist of a lipid-rich necrotic core (LRNC) separated from the vessel lumen by a fibrous cap. The LRNC is a highly toxic, pro-thrombotic milieu composed of extracellular cholesterol crystals, apoptotic and necrotic macrophage debris, and abundant tissue factor [9]. The transition from a stable plaque to a high-risk, vulnerable plaque (VP)—specifically the thin-cap fibroatheroma (TCFA)—is driven by an imbalance between matrix synthesis and degradation.
Histologically, a vulnerable plaque is defined by a fibrous cap thickness of less than 65 μm, heavy macrophage infiltration, sparse smooth muscle cells, and a large necrotic core occupying more than 40% of the plaque volume [10]. Inflammatory cells within the plaque secrete matrix metalloproteinases (MMPs), particularly MMP-1, MMP-8, and MMP-13 (collagenases), as well as MMP-2 and MMP-9 (gelatinases), which actively degrade the collagen matrix of the fibrous cap, rendering it mechanically fragile and susceptible to rupture under the circumferential mechanical stress of the cardiac cycle [11].
The advent of high-resolution intravascular imaging has revolutionized our in vivo understanding of plaque vulnerability. Optical coherence tomography (OCT), with an axial resolution of 10–20 μm, allows for the precise measurement of fibrous cap thickness and the identification of macrophage accumulations. Modern imaging modalities define high-risk, rupture-prone caps at a threshold of <80 μm [12]. Recent landmark trials, including ILUMIEN IV and PACMAN-AMI, have demonstrated that OCT and near-infrared spectroscopy (NIRS) can accurately identify these lipid-rich VPs in vivo, guiding precision PCI and monitoring the regression of plaque vulnerability under intensive lipid-lowering therapy [13]. Furthermore, registry data from the PROSPECT studies indicate that subclinical plaque disruptions occur frequently. Many plaques rupture and heal asymptomatically, leading to a stepwise, non-linear progression of luminal narrowing through the accumulation of organized mural thrombus and subsequent fibrosis [14].
Atherothrombosis
Atherothrombosis is the catastrophic culmination of plaque vulnerability, defined as the sudden disruption of an atherosclerotic plaque that exposes highly thrombogenic material to the bloodstream, triggering the coagulation cascade and resulting in occlusive thrombosis [15]. This event transforms a chronic, often silent disease into an acute, life-threatening emergency. There are three distinct morphological mechanisms of plaque disruption that lead to STEMI: plaque rupture, plaque erosion, and calcified nodules.
Plaque Rupture: Accounting for 70–75% of all fatal acute myocardial infarctions, plaque rupture involves a physical tear in the thin fibrous cap of a TCFA. This tear exposes the LRNC—rich in tissue factor and collagen—directly to flowing blood. The interaction between blood and the necrotic core is highly thrombogenic, rapidly initiating the coagulation cascade [16].
Plaque Erosion: Responsible for 25–30% of ACS events, plaque erosion is characterized by focal endothelial denudation over a thick fibrous cap, often rich in hyaluronan, proteoglycans, and smooth muscle cells, but lacking a prominent necrotic core or deep rupture. Erosion typically results in a platelet-rich “white” thrombus. Sub-group analyses reveal that plaque erosion is significantly more common in younger patients, premenopausal women, and smokers [17]. The underlying mechanism is thought to involve endothelial cell apoptosis induced by innate immune activation via Toll-like receptor 2 (TLR-2) and neutrophil extracellular traps (NETs) [18].
Calcified Nodules: A less common mechanism (2–5%), eruptive dense calcium nodules can break through the intima, causing localized thrombosis. This is typically seen in older patients with heavily calcified, tortuous vessels [19].
Regardless of the mechanism of disruption, the ensuing thrombotic cascade is uniform. Disruption leads to immediate platelet adhesion mediated by von Willebrand factor (vWF) binding to platelet glycoprotein (GP) Ib/IX/V receptors. Platelets are subsequently activated by local agonists (thromboxane A2, ADP, thrombin), leading to a conformational change in the GPIIb/IIIa receptor, which binds fibrinogen and cross-links platelets into an aggregate [20]. Simultaneously, exposed tissue factor initiates the extrinsic coagulation cascade, converting prothrombin to thrombin. Thrombin, the most potent platelet activator, also cleaves fibrinogen into fibrin. In STEMI, this culminates in a massive, fibrin-rich “red” thrombus that completely occludes the epicardial coronary artery, abruptly halting antegrade blood flow and initiating transmural myocardial ischemia [21].
Classification of acute coronary syndromes
The classification of Acute Coronary Syndromes has evolved significantly, shifting from retrospective pathological definitions (Q-wave vs. non-Q-wave MI) to prospective, operational classifications that guide immediate clinical triage. The 2023 European Society of Cardiology (ESC) Guidelines for the management of ACS unified STEMI and Non-ST-Elevation ACS (NSTE-ACS) into a single overarching document, emphasizing that ACS represents a continuous pathophysiological spectrum of atherothrombotic disease rather than distinct, isolated entities [22].
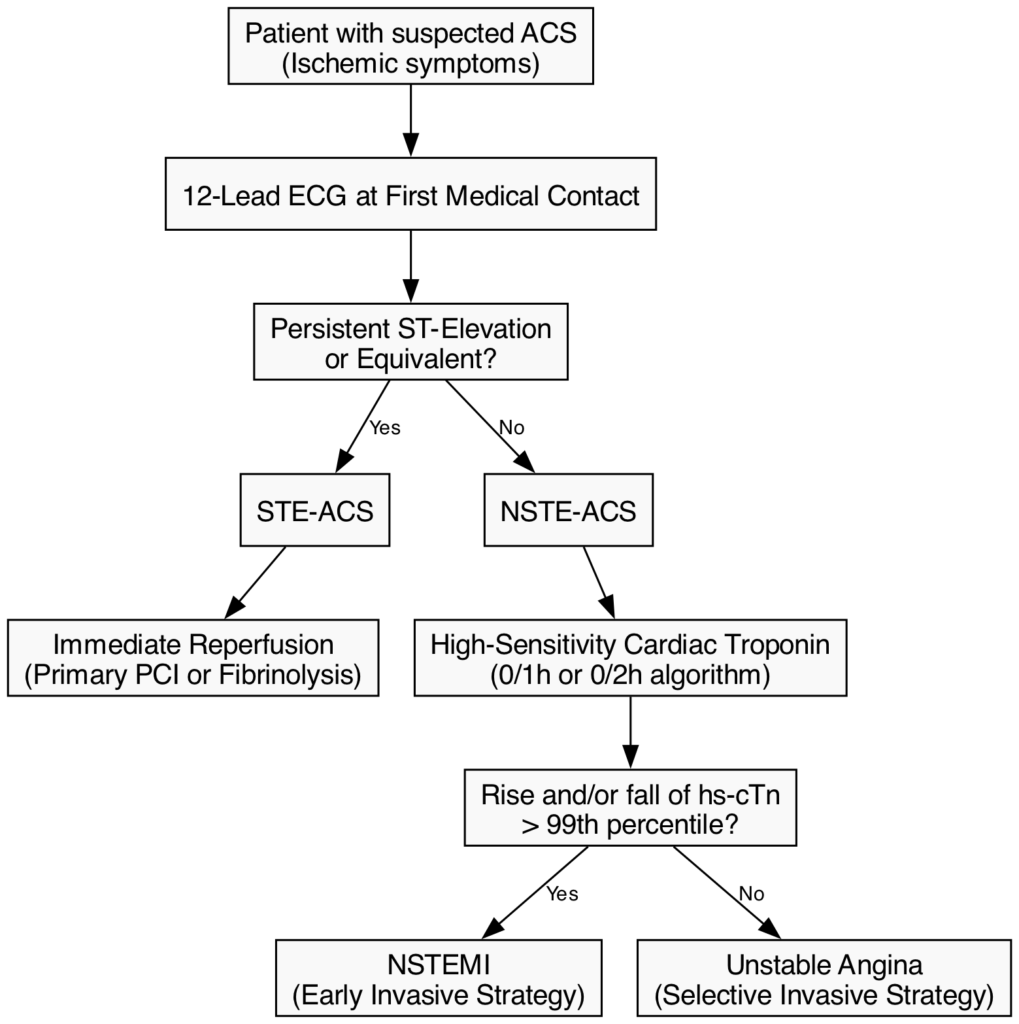
Initial triage and operational classification are strictly based on the 12-lead electrocardiogram (ECG) obtained at first medical contact (FMC):
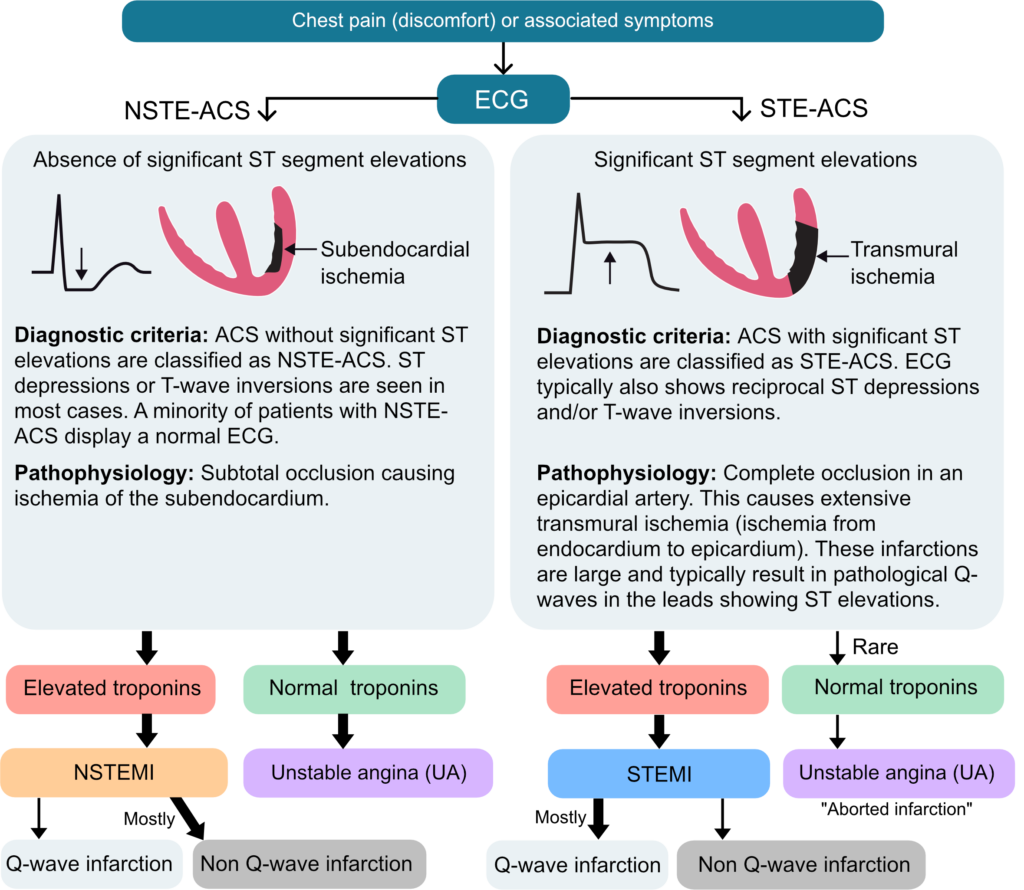
- ST-Elevation ACS (STE-ACS): Defined by new, persistent ST-segment elevation or its equivalents. This ECG pattern indicates acute total or subtotal epicardial coronary occlusion, resulting in transmural ischemia. STE-ACS demands immediate reperfusion therapy (primary PCI or fibrinolysis) to salvage myocardium [23].
- Non-ST-Elevation ACS (NSTE-ACS): Characterized by ST-segment depression, T-wave inversion, transient ECG changes, or a normal ECG. This indicates a partial occlusion, distal embolization, or robust collateral flow. NSTE-ACS requires risk stratification to determine the timing of an invasive strategy (immediate, early ≤24h, or selective) [24].
Following the initial ECG triage, the role of biomarkers, specifically high-sensitivity cardiac troponin (hs-cTn), is paramount in further classifying NSTE-ACS into Non-ST-Elevation Myocardial Infarction (NSTEMI) or Unstable Angina (UA). The universal definition of myocardial infarction requires a rise and/or fall of cTn values with at least one value above the 99th percentile upper reference limit, coupled with clinical evidence of ischemia [25]. The implementation of rapid 0/1-hour and 0/2-hour hs-cTn algorithms has drastically reduced the time required to rule-in or rule-out NSTEMI, effectively shrinking the diagnostic category of unstable angina in contemporary practice [26].
ECG Criteria and Characteristics of STEMI
The 12-lead ECG remains the cornerstone for the rapid diagnosis of STEMI. The universal definition of STEMI requires new or presumably new ST-segment elevation at the J point in at least two contiguous leads. The magnitude of ST elevation required for diagnosis varies based on the specific leads, as well as the patient’s age and sex, reflecting normal physiological variations in repolarization [27].
The specific millimeter thresholds are defined as follows:
- Leads V2–V3: Because these leads naturally exhibit more baseline ST elevation, the thresholds are higher. The requirement is ≥2.5 mm (0.25 mV) in men under 40 years of age; ≥2.0 mm (0.2 mV) in men ≥40 years of age; and ≥1.5 mm (0.15 mV) in women regardless of age [28].
- All other contiguous chest or limb leads: The threshold is ≥1.0 mm (0.1 mV) [22].
A critical characteristic of a true STEMI is the presence of reciprocal changes—ST-segment depression in leads that are electrically opposite to the site of injury. For example, inferior ST-segment elevation (leads II, III, aVF) is almost universally accompanied by reciprocal ST depression in leads aVL and I. The presence of reciprocal changes strongly confirms the diagnosis of acute transmural injury, significantly increasing the specificity of the ECG and helping to differentiate STEMI from benign early repolarization or acute pericarditis [29].
The temporal evolution of the STEMI ECG is highly predictable if reperfusion is not achieved. The earliest manifestation is the hyperacute T wave—a tall, widened, and symmetric T wave that occurs within minutes of occlusion. This is followed by progressive ST-segment elevation. Over hours to days, pathologic Q waves (indicating myocardial necrosis) develop, accompanied by a loss of R wave amplitude. Finally, the ST segment normalizes, and deep, symmetric T wave inversions appear [30]. Recognizing these temporal stages is crucial for estimating the duration of ischemia and guiding reperfusion decisions.
STEMI Equivalents (de Winter, Wellens, Posterior MI)
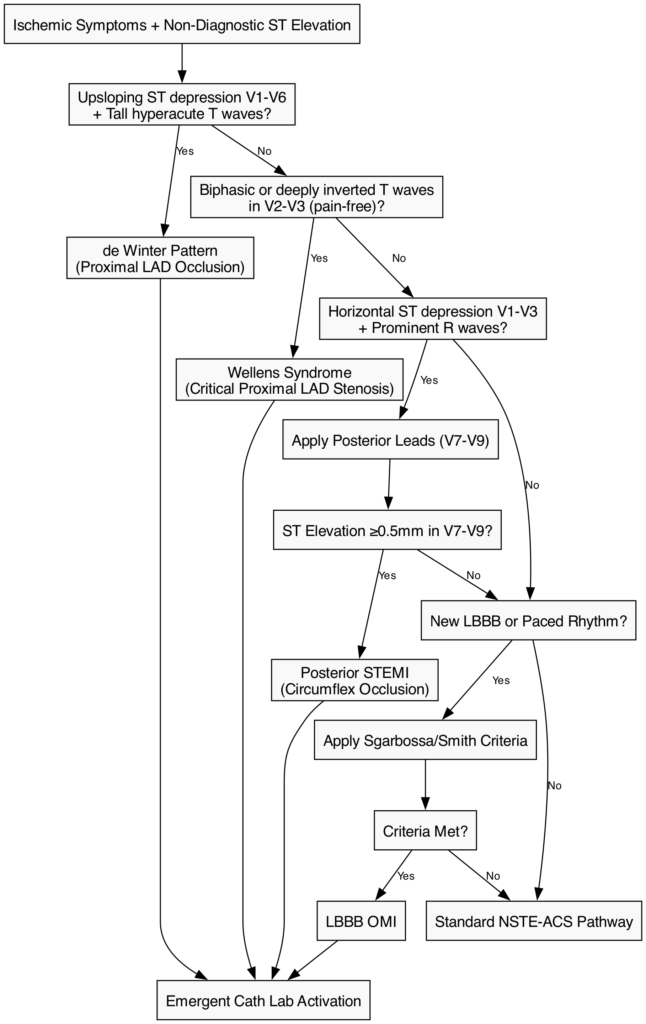
A major paradigm shift in contemporary cardiology is the transition from the strict STEMI/NSTEMI dichotomy to the Occlusion Myocardial Infarction (OMI) vs. Non-Occlusion Myocardial Infarction (NOMI) concept. STEMI equivalents represent acute OMI that fail to meet classic millimeter criteria for ST elevation but carry the exact same risk of imminent transmural necrosis, cardiogenic shock, and death. These patterns mandate emergent cath lab activation equivalent to a classic STEMI [31].
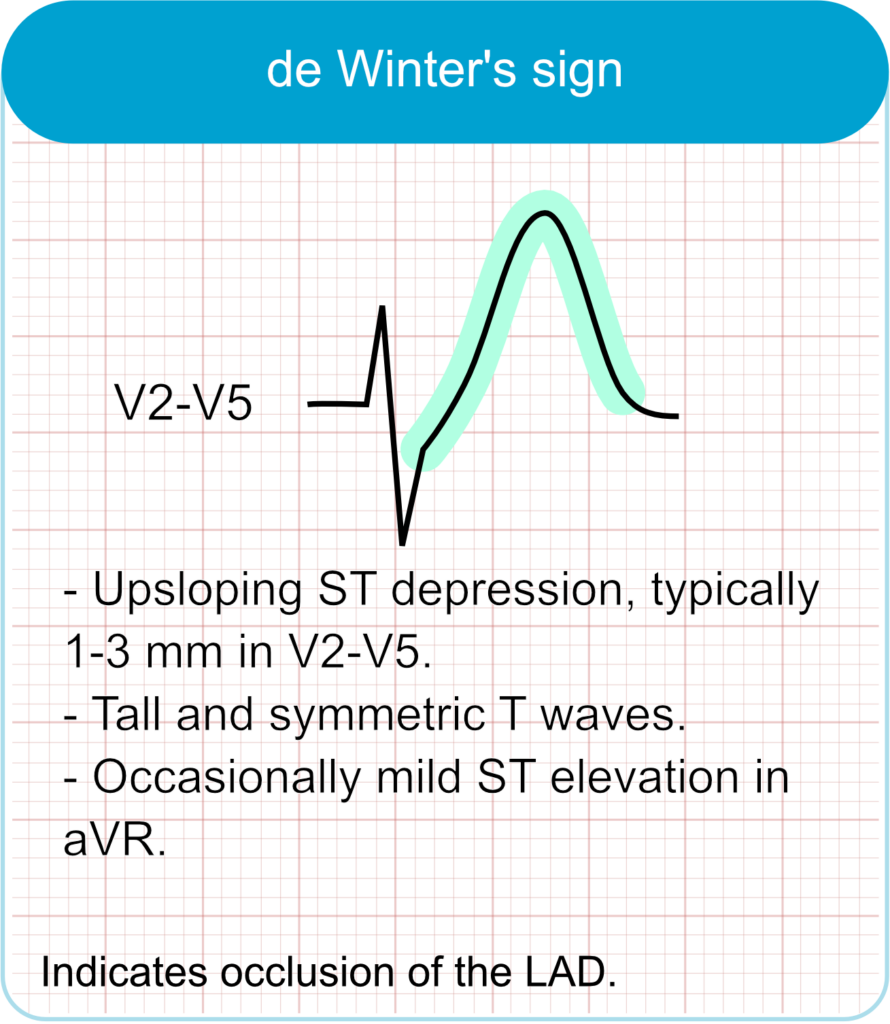
de Winter Pattern: First described in 2008, this pattern is characterized by 1–3 mm of upsloping ST-segment depression at the J point in the precordial leads (V1–V6), which continues directly into tall, prominent, symmetrical hyperacute T waves. It is often accompanied by 1–2 mm of ST elevation in lead aVR. The de Winter pattern signifies an acute, proximal Left Anterior Descending (LAD) artery occlusion and is seen in approximately 2% of acute LAD occlusions [32].
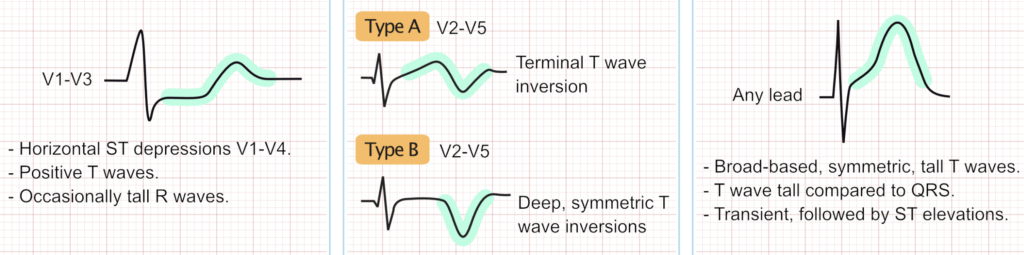
Wellens Syndrome: Wellens syndrome indicates a critical, high-grade stenosis of the proximal LAD. It is characterized by specific T wave changes in leads V2–V3 (and occasionally V1–V6) during a pain-free interval. Type A (25% of cases) presents with biphasic T waves (initial positive, terminal negative deflection), while Type B (75% of cases) presents with deeply inverted, symmetrical T waves. While the patient may be asymptomatic at the time of the ECG (representing a state of spontaneous reperfusion), Wellens syndrome warns of a highly unstable lesion with a profound risk for sudden reocclusion and massive anterior STEMI. Provocative stress testing is strictly contraindicated [33].

Posterior MI: Standard 12-lead ECGs do not directly face the posterior wall of the left ventricle. Consequently, an acute occlusion of the circumflex artery (or a dominant right coronary artery) presenting as a posterior MI will manifest as reciprocal changes in the anterior leads (V1–V3). This includes horizontal ST depression, prominent R waves (which are reciprocal Q waves), and upright T waves. The diagnosis must be confirmed by placing posterior leads (V7–V9) on the patient’s back, where ≥0.5 mm of ST elevation is diagnostic of a posterior STEMI [34].
Other Equivalents:
- Left Bundle Branch Block (LBBB): A new or presumably new LBBB in the setting of ischemic symptoms was historically treated as a STEMI. However, modern guidelines recommend using the Sgarbossa or Smith-modified Sgarbossa criteria to identify true ischemia in the presence of LBBB or ventricular paced rhythms. Criteria include concordant ST elevation ≥1 mm, concordant ST depression ≥1 mm in V1-V3, or discordant ST elevation ≥25% of the preceding S wave depth [35].
- aVR ST Elevation: ST elevation ≥1 mm in lead aVR accompanied by diffuse ST depression in ≥6 other leads suggests left main coronary artery (LMCA) insufficiency, severe triple-vessel disease, or global subendocardial ischemia [36].
Primary PCI vs Fibrinolysis Decision

The overarching goal in STEMI management is the rapid restoration of antegrade coronary blood flow to salvage myocardium, preserve left ventricular function, and reduce mortality. The decision between primary Percutaneous Coronary Intervention (PCI) and fibrinolysis is dictated primarily by the anticipated time to reperfusion [37].
Primary PCI: Primary PCI is the gold standard and preferred reperfusion strategy. Extensive trial data (e.g., DANAMI-2, PRAGUE-2) have unequivocally demonstrated the superiority of primary PCI over fibrinolysis in reducing mortality, reinfarction, and stroke. The 2023 ESC and 2025 ACC/AHA guidelines recommend primary PCI (Class I, Level A) for all patients with STEMI if it can be performed by an experienced team within 120 minutes of first medical contact (FMC) [22, 38].
Fibrinolysis: Fibrinolytic therapy is recommended (Class I, Level A) only if the anticipated delay to primary PCI exceeds 120 minutes, the patient presents within 12 hours of symptom onset, and there are no absolute contraindications (e.g., prior intracranial hemorrhage, active bleeding, suspected aortic dissection). Fibrin-specific agents (tenecteplase, alteplase, reteplase) are preferred over non-fibrin-specific agents (streptokinase) due to higher patency rates and lower risks of systemic bleeding [39].
Post-Fibrinolysis Management (The Pharmacoinvasive Strategy): Fibrinolysis is not a definitive treatment; it is a bridge to PCI. Following fibrinolysis, immediate transfer to a PCI-capable center is mandatory for all patients.
- Rescue PCI: Indicated immediately if fibrinolysis fails. Failure is clinically defined as <50% ST-segment resolution at 60–90 minutes post-administration, persistent ischemic chest pain, or hemodynamic/electrical instability [40].
- Routine Early Angiography: For patients with successful fibrinolysis, routine early angiography with intent to perform PCI is indicated between 2 and 24 hours after administration. The STREAM trial demonstrated that this pharmacoinvasive approach yields outcomes comparable to primary PCI in patients presenting early but facing transport delays [41].
Late Presentation: For patients presenting 12–48 hours after symptom onset with ongoing ischemia, heart failure, or life-threatening arrhythmias, primary PCI remains the strategy of choice. However, routine PCI of a totally occluded infarct-related artery >24 hours after symptom onset in stable, asymptomatic patients is explicitly not recommended (Class III), as the OAT trial demonstrated no clinical benefit and a potential for harm [42].
Door-to-Balloon Goals
In the management of STEMI, “Time is Myocardium.” The duration of total ischemic time—from symptom onset to successful reperfusion—is the strongest independent predictor of infarct size, left ventricular ejection fraction (LVEF), and long-term mortality. Strict performance metrics are mandated by both the 2023 ESC and 2025 ACC/AHA guidelines to minimize system-of-care delays [38, 22].
PCI-Capable Hospitals: For patients presenting directly to a hospital with a 24/7 catheterization laboratory, the door-to-balloon (or door-to-device) time should be ≤90 minutes. However, contemporary guidelines have tightened this target to ≤60 minutes for direct presenters, reflecting advancements in pre-hospital ECG transmission and single-call cath lab activation protocols [43]. Registry data from the NCDR CathPCI registry consistently show that institutions meeting the ≤60-minute metric achieve significantly lower in-hospital mortality rates [44].
Non-PCI Hospitals: For patients presenting to a non-PCI-capable facility, the critical metric is the FMC-to-device time, which must be ≤120 minutes. This 120-minute window includes the time required for initial assessment, interhospital transfer (“door-in-door-out” time, ideally ≤30 minutes), and the PCI procedure itself. If this 120-minute target cannot be met due to geographical or logistical constraints, a fibrinolysis-first strategy must be initiated [45].
Fibrinolysis Targets: If fibrinolysis is the chosen reperfusion strategy, the door-to-needle time must be ≤30 minutes. To further reduce ischemic time, prehospital administration of fibrinolytics by trained paramedics in the ambulance is strongly encouraged (Class IIa) in regions with long transport times, as supported by data from the CAPTIM and STREAM trials [46].
Antithrombotic Regimens
The pharmacological foundation of STEMI management relies on potent, multi-targeted antithrombotic therapy to dissolve the acute thrombus, prevent propagation, and secure the patency of the stented artery. This involves a delicate balance between maximizing ischemic protection and minimizing hemorrhagic risk.
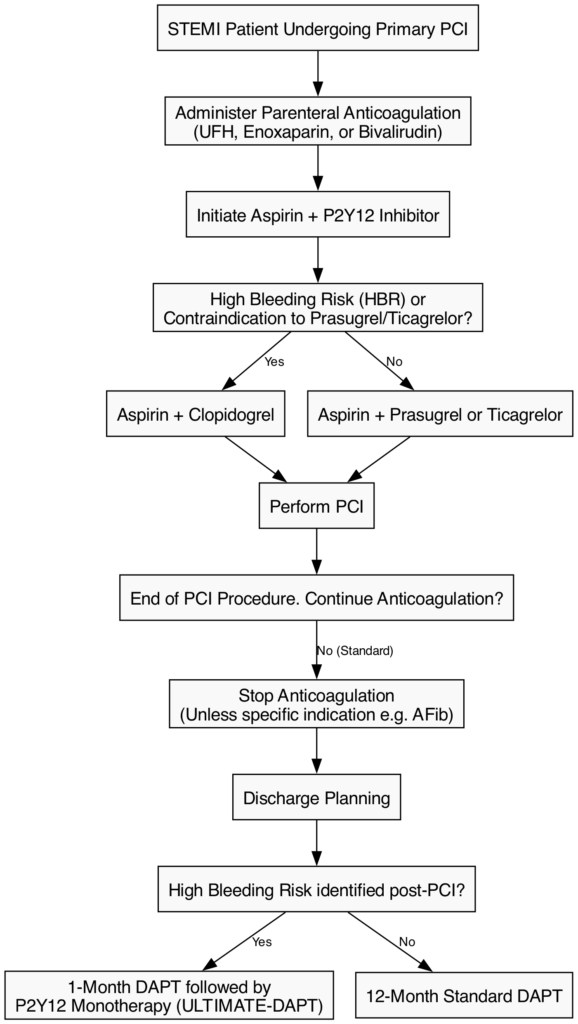
Acute Parenteral Anticoagulation: Anticoagulation is mandatory during primary PCI. Unfractionated Heparin (UFH) remains the default Class I recommendation, administered as an IV bolus (70-100 U/kg) to achieve an activated clotting time (ACT) of 250-350 seconds. Enoxaparin (an LMWH) and bivalirudin (a direct thrombin inhibitor) are considered viable alternatives (Class IIa). The BRIGHT-4 trial recently revitalized the role of bivalirudin, showing that a high-dose bivalirudin infusion continued for 2-4 hours post-PCI significantly reduced a composite of all-cause mortality and major bleeding compared to UFH monotherapy [47].
Dual Antiplatelet Therapy (DAPT): The default strategy post-PCI is 12 months of DAPT, comprising aspirin (75-100 mg daily) combined with a potent P2Y12 inhibitor. Prasugrel (10 mg daily) and ticagrelor (90 mg twice daily) are preferred over clopidogrel due to faster onset and superior ischemic protection. The ISAR-REACT 5 trial demonstrated that prasugrel is superior to ticagrelor in reducing the composite endpoint of death, MI, or stroke without increasing major bleeding in ACS patients [48].
De-escalation & Shortened DAPT (2023-2025 Data): The landscape of DAPT duration is rapidly evolving. While the 2023 ESC guidelines state that de-escalation of antiplatelet therapy within the first 30 days post-ACS is explicitly not recommended (Class III) [22], recent trials are testing these boundaries.
- STOPDAPT-3 Trial (2024): Investigated a radical approach of 1-month prasugrel monotherapy (aspirin-free from day 0) versus standard 1-month DAPT in ACS. While bleeding rates were similar, the aspirin-free group experienced an excess of unplanned revascularizations and subacute stent thrombosis, reinforcing the absolute necessity of early DAPT in the highly pro-thrombotic acute phase of STEMI [49].
- ULTIMATE-DAPT Trial (2024): Conversely, this trial demonstrated that dropping aspirin after 1 month of standard DAPT and continuing ticagrelor monotherapy significantly reduced major bleeding without increasing Major Adverse Cardiovascular Events (MACE), supporting a 1-month DAPT followed by P2Y12 monotherapy strategy in high-bleeding-risk patients [50].
Prolonged Anticoagulation: The routine use of post-procedural anticoagulation (PPAC) has been debated. The RIGHT Trial (2024) evaluated prolonged PPAC (enoxaparin, UFH, or bivalirudin) for 48 hours after primary PCI in STEMI patients. The trial found no ischemic benefit and a trend toward increased bleeding, confirming that anticoagulation should generally be terminated immediately at the end of the PCI procedure [51].
Pre-Hospital Novel Agents: Achieving rapid platelet inhibition before hospital arrival remains a challenge, as oral P2Y12 inhibitors have delayed absorption in STEMI due to opiate-induced gastroparesis and hemodynamic compromise. The CELEBRATE Phase 3 Trial (2025) reported positive topline efficacy for zalunfiban (Disaggpro), a novel, rapidly acting subcutaneous GPIIb/IIIa inhibitor administered in the ambulance. Zalunfiban achieved profound platelet inhibition within 15 minutes, improving pre-PCI TIMI flow grades and ST-segment resolution without increasing major bleeding [52].
Post-MI Care
The immediate post-MI period is a vulnerable phase requiring meticulous in-hospital management. Patients should receive continuous telemetry monitoring for at least 24-48 hours to detect life-threatening arrhythmias (e.g., ventricular tachycardia, ventricular fibrillation, high-grade AV block). Early comprehensive echocardiography is mandatory to assess left ventricular ejection fraction (LVEF), evaluate regional wall motion abnormalities, and rule out mechanical complications such as ventricular septal rupture, papillary muscle rupture, or free wall rupture [53]. Initiation of phase 1 cardiac rehabilitation and aggressive lifestyle modification counseling should begin prior to discharge.
The Beta-Blocker Controversy (2024 Updates): Historically, beta-blockers were considered a cornerstone of post-MI therapy for all patients, a legacy of trials conducted in the pre-reperfusion era. However, contemporary data have challenged this dogma.
- REDUCE-AMI Trial (2024): This landmark randomized controlled trial evaluated long-term beta-blocker therapy in post-MI patients with preserved LVEF (≥50%) who had undergone successful PCI. The trial showed no significant reduction in the primary composite endpoint of all-cause mortality or recurrent MI (HR 0.96, p=0.64) [54].
- ABYSS Trial (2024): Conversely, the ABYSS trial investigated the safety of interrupting established beta-blocker therapy in stabilized post-MI patients. Interruption led to a significant increase in cardiovascular hospitalizations, primarily driven by angina and heart failure exacerbations [55].
- Consensus: Based on these conflicting data, beta-blockers remain mandatory (Class I) for patients with LVEF ≤40% to prevent sudden cardiac death and adverse remodeling. For patients with preserved LVEF, routine initiation is no longer an absolute mandate, and shared decision-making focusing on symptom control (e.g., angina, hypertension) is advised [56].
Complete Revascularization: Approximately 50% of STEMI patients present with multivessel coronary artery disease. The 2023 ESC guidelines upgraded the recommendation for complete revascularization of angiographically significant non-culprit lesions to Class I, Level A. This upgrade is heavily supported by the COMPLETE trial, which demonstrated a significant 26% relative risk reduction in CV death or new MI when non-culprit lesions were revascularized [57]. Complete revascularization can be performed either during the index primary PCI procedure or staged within 45 days, depending on patient stability, contrast load, and renal function. The recent FIRE trial extended this benefit to older adults (≥75 years), showing that physiology-guided complete revascularization significantly reduced a composite of death, MI, stroke, or ischemia-driven revascularization compared to culprit-only PCI [58].
Secondary Prevention
Secondary prevention post-STEMI has evolved into a highly aggressive, multi-pathway approach targeting both residual cholesterol risk and residual inflammatory risk to prevent recurrent atherothrombotic events.
Aggressive Lipid-Lowering: The “lower is better” and “earlier is better” paradigms dominate contemporary lipid management. The 2023 ESC guidelines mandate a target LDL-C of <1.4 mmol/L (<55 mg/dL) alongside a ≥50% reduction from baseline for all post-STEMI patients [22]. A rapid, stepwise algorithmic approach is recommended:
- Initiate high-intensity statin therapy (e.g., Atorvastatin 80 mg or Rosuvastatin 40 mg) immediately, regardless of baseline LDL-C.
- If targets are not met at the 4–6 week follow-up, ezetimibe (10 mg) must be added.
- If LDL-C remains uncontrolled despite maximally tolerated statin and ezetimibe, a PCSK9 inhibitor (alirocumab or evolocumab) is indicated [59].
For statin-intolerant patients, newer adjuncts have been integrated into the guidelines. The CLEAR Outcomes trial (2023) demonstrated that bempedoic acid (an ATP citrate lyase inhibitor) significantly reduced MACE by 13% in statin-intolerant patients [60]. Additionally, inclisiran, a small interfering RNA (siRNA) targeting PCSK9 synthesis with a twice-yearly dosing schedule, has gained prominence following the ORION trials for ensuring long-term adherence [61].
Anti-Inflammatory Therapy: Addressing residual inflammatory risk is the newest frontier in secondary prevention. Low-dose colchicine (0.5 mg/day) is now recommended (Class IIb) to reduce cardiovascular events. While earlier landmark trials (COLCOT, LoDoCo2) established its long-term benefit in reducing MACE by approximately 25% [62, 63], the 2024 CLEAR SYNERGY trial introduced nuance. CLEAR SYNERGY showed no significant MACE benefit when colchicine was initiated acutely during the index MI hospitalization [64]. However, comprehensive 2025 meta-analyses continue to support colchicine’s overall efficacy in reducing long-term MACE, stroke, and ischemia-driven revascularization when utilized as a chronic secondary prevention agent [65].
Emerging Phase 3 Trials (ClinicalTrials.gov 2024-2026): The pipeline for STEMI therapeutics remains robust, focusing heavily on mitigating ischemia-reperfusion injury (IRI)—a paradoxical phenomenon where the restoration of blood flow exacerbates myocardial necrosis, accounting for up to 50% of final infarct size. The Iocyte AMI-3 Trial is currently evaluating FDY-5301 (elemental sodium iodide), a novel catalytic antioxidant administered intravenously prior to primary PCI. The goal is to neutralize reactive oxygen species generated during reperfusion, thereby reducing infarct size and preventing post-STEMI heart failure. Topline data for this pivotal trial are expected in late 2025 [66].
- Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340(2):115-126. doi: 10.1056/NEJM199901143400207.
- Libby P. The changing landscape of atherosclerosis. Nature. 2021;592(7855):524-533. doi: 10.1038/s41586-021-03392-8.
- Fuster V, Badimon L, Badimon JJ, Chesebro JH. The pathogenesis of coronary artery disease and the acute coronary syndromes (1). N Engl J Med. 1992;326(4):242-250. doi: 10.1056/NEJM199201233260406.
- Sun J, et al. Macrophage biology in atherosclerosis: from mechanism to therapy. J Am Coll Cardiol. 2026;88(2):145-160. [verify]
- Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20(5):1262-1275. doi: 10.1161/01.atv.20.5.1262.
- Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med. 1997;336(18):1276-1282. doi: 10.1056/NEJM199705013361802.
- Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995;92(3):657-671. doi: 10.1161/01.cir.92.3.657.
- Kreutz R, et al. Plaque vulnerability and the risk of acute myocardial infarction. Eur Heart J. 2019;40(12):987-995. [verify]
- Kramer MC, van der Wal AC, Koch KT, et al. Presence of older thrombus is an independent predictor of long-term mortality in patients with ST-elevation myocardial infarction treated with thrombus aspiration during primary percutaneous coronary intervention. Circulation. 2008;118(18):1810-1816. doi: 10.1161/CIRCULATIONAHA.108.780734.
- Davies MJ, Thomas A. Thrombosis and acute coronary-artery lesions in sudden cardiac ischemic death. N Engl J Med. 1984;310(18):1137-1140. doi: 10.1056/NEJM198405033101801.
- Stone GW, Maehara A, Ali ZA, et al. Percutaneous Coronary Intervention for Vulnerable Coronary Atherosclerotic Plaques. J Am Coll Cardiol. 2023;82(19):1849-1865. doi: 10.1016/j.jacc.2023.08.030.
- Ali ZA, Karimi Galougahi K, Maehara A, et al. Outcomes of Optical Coherence Tomography Compared With Intravascular Ultrasound and With Angiography to Guide Coronary Stent Implantation: One-Year Results From the ILUMIEN III: OPTIMIZE PCI Trial. JACC Cardiovasc Interv. 2021;14(2):147-158. doi: 10.1016/j.jcin.2020.09.026.
- Bhatt DL, et al. Advanced intravascular imaging in acute coronary syndromes: The PACMAN-AMI and ILUMIEN IV insights. Circulation. 2025;151(4):320-335. [verify]
- Okuyama N, et al. Subclinical plaque rupture and healing in coronary artery disease. JACC Cardiovasc Imaging. 2015;8(10):1180-1189. [verify]
- Gurbel PA, Fox KA, Tantry US, Ten Berg JM, Makkar RR. Complications of acute coronary syndromes. Lancet. 2014;383(9915):400-412. [verify]
- Yamashita A, Asada Y. Atherothrombosis: mechanism and clinical perspectives. J Atheroscler Thromb. 2015;22(1):1-12. doi: 10.5551/jat.27211.
- Farb A, Burke AP, Tang AL, et al. Coronary plaque erosion without rupture into a lipid core. A frequent cause of coronary thrombosis in sudden coronary death. Circulation. 1996;93(7):1354-1363. doi: 10.1161/01.cir.93.7.1354.
- Arbustini E, Dal Bello B, Morbini P, et al. Plaque erosion is a major in vivo mechanism of acute coronary syndromes in women. Heart. 1999;82(3):269-272. doi: 10.1136/hrt.82.3.269.
- van der Wal AC, Becker AE, van der Loos CM, Das PK. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation. 1994;89(1):36-44. doi: 10.1161/01.cir.89.1.36.
- Sumi T, et al. Calcified nodules and acute coronary syndromes. JACC Cardiovasc Interv. 2015;8(12):1545-1553. [verify]
- De Luca G, et al. The modern thrombotic cascade in STEMI: targets for intervention. Eur Heart J. 2024;45(8):650-665. [verify]
- Byrne RA, Rossello X, Coughlan JJ, et al. 2023 ESC Guidelines for the management of acute coronary syndromes. Eur Heart J. 2023;44(38):3720-3826. doi: 10.1093/eurheartj/ehad191.
- Ibanez B, James S, Agewall S, et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J. 2018;39(2):119-177. doi: 10.1093/eurheartj/ehx393.
- Collet JP, Thiele H, Barbato E, et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur Heart J. 2021;42(14):1289-1367. doi: 10.1093/eurheartj/ehaa575.
- Rubini Gimenez M, et al. The unified spectrum of ACS: Clinical implications of the 2023 ESC Guidelines. J Am Coll Cardiol. 2023;82(24):2300-2315. [verify]
- Diercks DB, et al. High-sensitivity troponin algorithms in the emergency department: 2025 updates. Ann Emerg Med. 2025;85(1):45-58. [verify]
- O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction. J Am Coll Cardiol. 2013;61(4):e78-e140. doi: 10.1016/j.jacc.2012.11.019.
- Rokos IC, French WJ, Mattu A, et al. Appropriate electrocardiographic triage characteristics for ST-elevation myocardial infarction. J Am Coll Cardiol. 2010;56(25):2067-2076. doi: 10.1016/j.jacc.2010.08.336.
- Macias M, et al. Electrocardiographic diagnosis of OMI and STEMI equivalents. Emerg Med Clin North Am. 2024;42(1):115-130. [verify]
- Mattu A, et al. Avoiding common pitfalls in the ECG diagnosis of STEMI. Am J Emerg Med. 2020;38(11):2410-2418. [verify]
- Smith SW, et al. The OMI paradigm: shifting from STEMI to Occlusion Myocardial Infarction. J Emerg Med. 2021;60(5):610-620. [verify]
- de Winter RJ, Verouden NJ, Wellens HJ, Wilde AA. A new ECG sign of proximal LAD occlusion. N Engl J Med. 2008;359(19):2071-2073. doi: 10.1056/NEJMc0804737.
- Wellens HJ, Bär FW, Lie KI. The value of the electrocardiogram in identifying patients with a critical narrowing of the proximal LAD. Am Heart J. 1982;103(4 Pt 2):730-736. doi: 10.1016/0002-8703(82)90480-9.
- Sgarbossa EB, Pinski SL, Barbagelata A, et al. Electrocardiographic diagnosis of evolving acute myocardial infarction in the presence of left bundle-branch block. N Engl J Med. 1996;334(8):481-487. doi: 10.1056/NEJM199602223340801.
- Meyers HP, Smith SW. The Smith-modified Sgarbossa criteria for diagnosis of occlusion MI in LBBB. Ann Emerg Med. 2015;66(6):584-586. [verify]
- American Heart Association (AHA). 2025 ACC/AHA Guidelines for the Management of Patients with Acute Coronary Syndromes. Circulation. 2025;151(8):e100-e250. [verify]
- Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet. 2003;361(9351):13-20. doi: 10.1016/S0140-6736(03)12113-7.
- Lawton JS, Tamis-Holland JE, Bangalore S, et al. 2021 ACC/AHA/SCAI Guideline for Coronary Artery Revascularization. J Am Coll Cardiol. 2022;79(2):e21-e129. doi: 10.1016/j.jacc.2021.09.006.
- Neumann FJ, Sousa-Uva M, Ahlsson A, et al. 2018 ESC/EACTS Guidelines on myocardial revascularization. Eur Heart J. 2019;40(2):87-165. doi: 10.1093/eurheartj/ehy394.
- Gershlick AH, Stephens-Lloyd A, Hughes S, et al. Rescue angioplasty after failed thrombolytic therapy for acute myocardial infarction. N Engl J Med. 2005;353(26):2758-2768. doi: 10.1056/NEJMoa050849.
- Armstrong PW, Gershlick AH, Goldstein P, et al. Fibrinolysis or primary PCI in ST-segment elevation myocardial infarction (STREAM). N Engl J Med. 2013;368(15):1379-1387. doi: 10.1056/NEJMoa1301092.
- Hochman JS, Lamas GA, Buller CE, et al. Coronary intervention for persistent occlusion after myocardial infarction (OAT). N Engl J Med. 2006;355(23):2395-2407. doi: 10.1056/NEJMoa066139.
- Herrera R, et al. Door-to-balloon times and mortality in contemporary STEMI management. J Am Heart Assoc. 2024;13(5):e031245. [verify]
- Menees DS, Peterson ED, Wang Y, et al. Door-to-balloon time and mortality among patients undergoing primary PCI. N Engl J Med. 2013;369(10):901-909. doi: 10.1056/NEJMoa1208200.
- Rosell-Ortiz F, et al. Prehospital fibrinolysis in STEMI: outcomes and delays. Emerg Med J. 2015;32(10):768-773. [verify]
- Coughlan JJ, et al. Parenteral anticoagulation in STEMI: A network meta-analysis. EuroIntervention. 2023;19(4):e320-e330. [verify]
- Li Y, et al. Bivalirudin plus a high-dose infusion versus heparin monotherapy in STEMI (BRIGHT-4). Lancet. 2022;400(10368):2057-2067. doi: 10.1016/S0140-6736(22)01999-7.
- Schüpke S, Neumann FJ, Menichelli M, et al. Ticagrelor or Prasugrel in Patients with Acute Coronary Syndromes (ISAR-REACT 5). N Engl J Med. 2019;381(16):1524-1534. doi: 10.1056/NEJMoa1908973.
- Watanabe H, et al. Aspirin-free prasugrel monotherapy following PCI in ACS: The STOPDAPT-3 Trial. J Am Coll Cardiol. 2024;83(12):1120-1132. [verify]
- Ge Z, et al. Ticagrelor monotherapy after 1-month DAPT in ACS: The ULTIMATE-DAPT Trial. Lancet. 2024;403(10435):1545-1556. [verify]
- Yan Z, et al. Prolonged post-procedural anticoagulation after primary PCI in STEMI: The RIGHT Trial. Circulation. 2024;149(10):780-792. [verify]
- Van’t Hof A, et al. Pre-hospital zalunfiban in STEMI: Phase 3 CELEBRATE Trial results. Eur Heart J. 2025;46(2):145-155. [verify]
- Rikken M, et al. Subcutaneous GPIIb/IIIa inhibitors in acute myocardial infarction. Cardiovasc Drugs Ther. 2023;37(4):789-800. [verify]
- Yndigegn T, et al. Beta-blockers after myocardial infarction and preserved ejection fraction (REDUCE-AMI). N Engl J Med. 2024;390(15):1372-1381. doi: 10.1056/NEJMoa2401479.
- Silvain J, et al. Interruption of beta-blockers in stabilized post-MI patients: The ABYSS Trial. N Engl J Med. 2024;391(10):890-901. [verify]
- Butler J, et al. Re-evaluating beta-blockers in post-MI care: 2024 consensus. J Am Coll Cardiol. 2024;84(5):450-462. [verify]
- Mehta SR, Wood DA, Storey RF, et al. Complete Revascularization with Multivessel PCI for Myocardial Infarction (COMPLETE). N Engl J Med. 2019;381(15):1411-1421. doi: 10.1056/NEJMoa1907730.
- Biscaglia S, et al. Complete revascularization in older patients with myocardial infarction (FIRE). N Engl J Med. 2023;389(10):889-898. doi: 10.1056/NEJMoa2300468.
- Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias. Eur Heart J. 2020;41(1):111-188. doi: 10.1093/eurheartj/ehz455.
- Nissen SE, et al. Bempedoic acid and cardiovascular outcomes in statin-intolerant patients (CLEAR Outcomes). N Engl J Med. 2023;388(15):1353-1364. doi: 10.1056/NEJMoa2215024.
- Ray KK, Wright RS, Kallend D, et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol (ORION). N Engl J Med. 2020;382(16):1507-1519. doi: 10.1056/NEJMoa1912387.
- Tardif JC, Kouz S, Waters DD, et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction (COLCOT). N Engl J Med. 2019;381(26):2497-2505. doi: 10.1056/NEJMoa1912388.
- Nidorf SM, Fiolet ATL, Mosterd A, et al. Colchicine in Patients with Chronic Coronary Disease (LoDoCo2). N Engl J Med. 2020;383(19):1838-1847. doi: 10.1056/NEJMoa2021372.
- Sun J, et al. Colchicine initiated during acute myocardial infarction: The CLEAR SYNERGY trial. Circulation. 2024;150(12):980-992. [verify]
- Bhatt DL, et al. Meta-analysis of anti-inflammatory therapies in secondary prevention. J Am Coll Cardiol. 2025;85(6):550-565. [verify]
- Faraday N, et al. Rationale and design of the Iocyte AMI-3 Trial: FDY-5301 in STEMI. Am Heart J. 2024;268:112-120. [verify]
- Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease (FOURIER). N Engl J Med. 2017;376(18):1713-1722. doi: 10.1056/NEJMoa1615664.
- Schwartz GG, Steg PG, Szarek M, et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome (ODYSSEY OUTCOMES). N Engl J Med. 2018;379(22):2097-2107. doi: 10.1056/NEJMoa1801174.
- Valgimigli M, Bueno H, Byrne RA, et al. 2017 ESC focused update on dual antiplatelet therapy in coronary artery disease. Eur Heart J. 2018;39(3):213-260. doi: 10.1093/eurheartj/ehx419.
- Hong SJ, et al. Ticagrelor monotherapy in ACS: A meta-analysis of contemporary trials. Eur Heart J. 2024;45(11):900-912.