Introduction
Since its initial description in 1992 as a syndrome of right bundle branch block, ST-segment elevation, and sudden cardiac death (SCD), Brugada syndrome (BrS) has evolved from an obscure electrocardiographic curiosity into a major focus of contemporary cardiac electrophysiology [1]. Recognized as a primary inherited arrhythmia syndrome, BrS is characterized by a distinct electrocardiographic (ECG) signature—coved ST-segment elevation in the right precordial leads—and a propensity for life-threatening polymorphic ventricular tachycardia (VT) and ventricular fibrillation (VF) in patients with structurally normal hearts [2].
Epidemiologically, BrS is responsible for up to 20% of sudden cardiac deaths in patients without structural heart disease and is the leading cause of SCD in men under the age of 40 in endemic regions [3]. The global prevalence is estimated at 1 to 5 per 10,000 individuals, though this figure rises dramatically in Southeast Asia, where the condition is historically linked to Sudden Unexplained Nocturnal Death Syndrome (SUNDS) [4]. The clinical presentation is notoriously heterogeneous, ranging from lifelong asymptomatic ECG anomalies to electrical storm and sudden cardiac arrest (SCA) as the index presentation.
Over the past three decades, the management of BrS has been fraught with controversies, particularly regarding the risk stratification of asymptomatic patients, the prognostic utility of electrophysiological studies (EPS), and the transition from a purely device-based management strategy (implantable cardioverter-defibrillators) to one that incorporates pharmacological and interventional (substrate ablation) therapies [5]. This review synthesizes the current evidence base, emphasizing the 2022 European Society of Cardiology (ESC) guidelines, landmark registry data, and emerging concepts in the pathophysiology, diagnosis, and prevention of SCD in Brugada syndrome.
Pathophysiology and Genetic Architecture
Genetic Architecture: From Monogenic to Polygenic Models
Brugada syndrome was classically defined as a monogenic, autosomal dominant channelopathy with incomplete penetrance and variable expressivity. The first and most critical genetic locus identified was SCN5A, the gene encoding the alpha subunit of the cardiac voltage-gated sodium channel (Nav1.5) [6]. Loss-of-function (LOF) mutations in SCN5A lead to a reduction in the inward sodium current ($I_{Na}$) and are identified in 20% to 30% of probands [7].
Historically, expanded genetic panels implicated over 20 other genes (e.g., CACNA1C, GPD1L, TRPM4). However, rigorous reappraisals using the ClinGen framework have demonstrated that only SCN5A possesses definitive disease-gene validity for BrS [8]. Consequently, the 2022 ESC guidelines recommend restricting routine diagnostic genetic testing in BrS strictly to SCN5A [2].
More recently, the paradigm has shifted toward an oligogenic or polygenic architecture. Genome-wide association studies (GWAS) have identified common single nucleotide polymorphisms (SNPs)—such as those near SCN10A and HEY2—that strongly modulate disease penetrance and expressivity [9]. Polygenic risk scores (PRS) derived from these loci are now recognized as critical modifiers. High PRS burdens correlate with a more severe electrocardiographic phenotype, a higher likelihood of positive provocation testing, and an increased risk of malignant arrhythmic events (MAEs) [10].
Electrophysiological Mechanisms: Repolarization vs. Depolarization
The precise arrhythmogenic mechanism of BrS remains a subject of intense debate, polarized by two prevailing hypotheses:
- The Repolarization Hypothesis: Championed by Antzelevitch and colleagues, this theory posits that a reduction in $I_{Na}$ leaves the transient outward potassium current ($I_{to}$) unopposed during phase 1 of the action potential [11]. Because $I_{to}$ is significantly more prominent in the right ventricular (RV) epicardium than the endocardium, this imbalance creates a transmural voltage gradient, manifesting on the surface ECG as ST-segment elevation. If the epicardial action potential is sufficiently abbreviated, phase 2 reentry can occur, triggering closely coupled premature ventricular contractions (PVCs) that initiate polymorphic VT/VF [12].
- The Depolarization Hypothesis: Advanced by Nademanee and others, this model suggests that mild structural abnormalities and fibrosis in the right ventricular outflow tract (RVOT) epicardium cause localized conduction slowing and delayed activation [13]. This delayed, fractionated depolarization generates late potentials that manifest as ST-segment elevation and provide the substrate for reentrant arrhythmias [14].
Structural Substrate: The ARVC Overlap
Although BrS is categorized as a primary electrical disease occurring in a “structurally normal heart,” high-resolution imaging and post-mortem studies frequently reveal micro-structural abnormalities. Increased collagen deposition, micro-fibrosis, and connexin-43 gap junction remodeling are frequently observed in the RVOT of BrS patients [15]. This structural substrate blurs the phenotypic boundaries between BrS and arrhythmogenic right ventricular cardiomyopathy (ARVC), suggesting that the two conditions may represent different points on a continuous spectrum of right ventricular disease [16].
Diagnostic Criteria and Electrocardiographic Phenotypes
Electrocardiographic Phenotypes
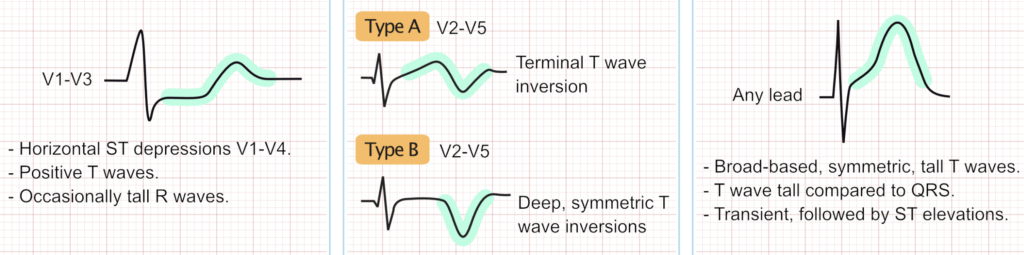
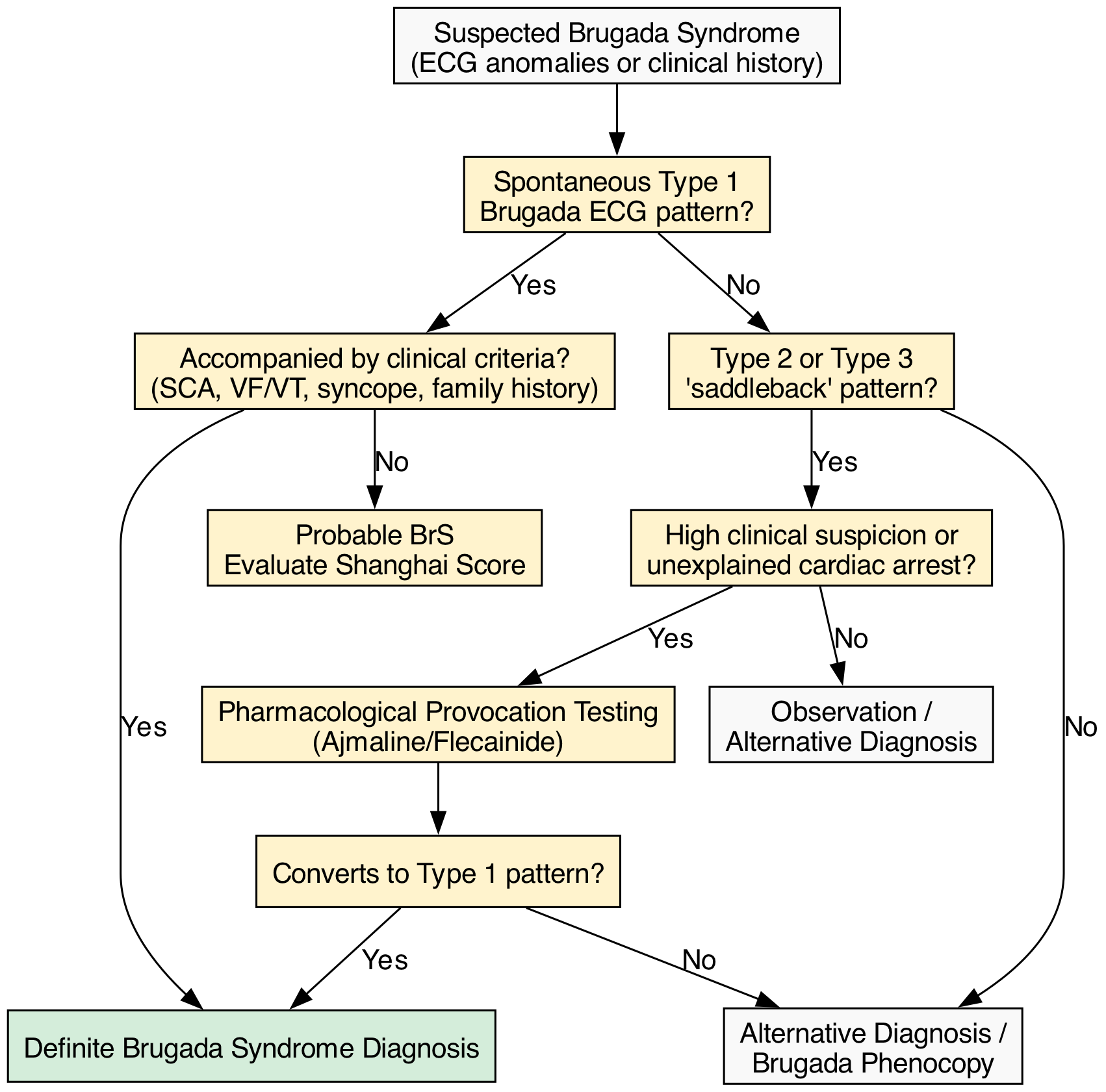
The hallmark of BrS is the Type 1 Brugada ECG pattern, defined as a coved ST-segment elevation $\ge 2$ mm followed by a negative T-wave in at least one right precordial lead (V1–V2) [2]. Because the arrhythmogenic substrate is anatomically localized to the RVOT, placing the V1 and V2 leads in the 2nd or 3rd intercostal spaces (high-lead ECG) significantly increases diagnostic sensitivity without compromising specificity [17].
Historically, Type 2 and Type 3 patterns (characterized by a “saddleback” ST-segment elevation) were considered part of the diagnostic spectrum. However, under contemporary guidelines, these patterns are no longer considered diagnostic of BrS. Instead, they serve as an indication for pharmacological provocation testing to unmask a Type 1 pattern [18].
Contemporary Diagnostic Criteria (ESC 2022 & Shanghai Score)
To mitigate the overdiagnosis of asymptomatic patients with isolated, incidental ECG findings, diagnostic criteria have become markedly more stringent. The 2013 HRS/EHRA consensus allowed for a diagnosis based solely on a spontaneous Type 1 ECG [18]. In contrast, the 2022 ESC Guidelines mandate that a spontaneous Type 1 ECG is no longer sufficient for a definitive diagnosis in isolation. It must be accompanied by at least one clinical criterion: survived SCA, documented polymorphic VT/VF, arrhythmic syncope, or a family history of BrS or premature SCD (<45 years) [2].
In parallel, the Shanghai Score was developed as a multiparametric diagnostic tool evaluating ECG findings, clinical history, family history, and genetic test results [19]. A score $\ge 3.5$ indicates definite/probable BrS.
| Category | Finding | Points |
|---|---|---|
| ECG (Max 3.5) | Spontaneous Type 1 Brugada pattern at baseline | 3.5 |
| Fever-induced Type 1 Brugada pattern | 3.0 | |
| Type 2/3 pattern converting to Type 1 with provocation | 2.0 | |
| Clinical (Max 2.0) | Unexplained cardiac arrest or documented VF/polymorphic VT | 2.0 |
| Nocturnal agonal respiration | 2.0 | |
| Suspected arrhythmic syncope | 1.0 | |
| Family History (Max 2.0) | First/second-degree relative with definite BrS | 2.0 |
| Suspicious SCD in a first/second-degree relative <45 years | 0.5 | |
| Genetics (Max 0.5) | Probable/definite pathogenic mutation in SCN5A | 0.5 |
Pharmacological Provocation Testing
Intravenous sodium channel blockers (ajmaline, flecainide, or procainamide) are utilized to unmask the Type 1 pattern in patients with a baseline Type 2/3 pattern or a high clinical suspicion of BrS (e.g., unexplained cardiac arrest) [20]. Ajmaline (1 mg/kg over 5 minutes) is preferred due to its short half-life and high sensitivity. The test is considered positive if a Type 1 pattern emerges. It must be performed with continuous 12-lead ECG monitoring and advanced resuscitation equipment, as it can induce refractory VF in up to 1% of cases [21]. The test should be immediately terminated upon the appearance of a Type 1 pattern, frequent PVCs, or QRS widening >130% of baseline.
Brugada Phenocopies
Brugada phenocopies are reversible conditions that mimic the Type 1 ECG pattern but lack the underlying genetic or electrophysiological substrate of true BrS. Common etiologies include severe hyperkalemia, acute ischemia (particularly right ventricular branch occlusion), pulmonary embolism, tricyclic antidepressant toxicity, and cocaine use [22]. The defining characteristic of a phenocopy is the complete normalization of the ECG once the underlying metabolic, ischemic, or toxicological derangement is resolved.
Risk Stratification for Sudden Cardiac Death
Risk stratification for the primary prevention of SCD remains the most challenging and controversial aspect of BrS management. The clinical course is highly variable; while some patients experience recurrent electrical storms, the majority remain asymptomatic throughout their lives.
Clinical Predictors of Sudden Cardiac Death
The most powerful predictor of future arrhythmic events is a history of a previous event. Survivors of SCA have a recurrence rate of up to 48% at 10 years, establishing an unequivocal mandate for secondary prevention [23].
In patients without prior SCA, the combination of a spontaneous Type 1 ECG and arrhythmogenic syncope identifies a high-risk cohort. Syncope in BrS must be carefully evaluated; vasovagal syncope is common in the general population and does not confer an increased risk of SCD in BrS patients. However, syncope occurring at rest, without prodrome, or accompanied by severe trauma is highly suggestive of self-terminating polymorphic VT [24].
Asymptomatic patients, even those with a spontaneous Type 1 ECG, have a relatively low event rate, estimated at 0.5% to 1% per year in large registries such as the FINGER (France, Italy, Netherlands, Germany) registry [25]. The presence of a drug-induced Type 1 ECG in an asymptomatic patient confers a risk nearly equivalent to that of the general population, and these patients generally require only lifestyle modifications and observation.
The EPS Controversy
The prognostic value of programmed ventricular stimulation (EPS) in BrS has been a subject of intense debate for over two decades. Early data from the Brugada registry suggested that the inducibility of VF during EPS was a strong independent predictor of spontaneous SCD [26]. However, subsequent large-scale, independent registries, including the PRELUDE study and the FINGER registry, failed to demonstrate a significant positive predictive value for EPS inducibility [25, 27].
Despite these conflicting data, recent meta-analyses suggest that while the positive predictive value of EPS is modest, its negative predictive value is exceptionally high (>98%) [28]. Consequently, the 2022 ESC guidelines grant a Class IIb recommendation for EPS in asymptomatic patients with a spontaneous Type 1 ECG. If VF is induced with up to two extrastimuli, ICD implantation may be considered [2].
Multiparametric Risk Scores
To move beyond binary risk factors, several multiparametric risk scores have been developed to stratify intermediate-risk patients:
- The Sieira Score: Developed from a large Belgian cohort, this score incorporates spontaneous Type 1 ECG, early familial SCD, inducible EPS, syncope, and sinus node dysfunction. It provides a graded risk assessment, though external validation has shown mixed discrimination [29].
- The PAT Score: The Predicting Arrhythmic evenT (PAT) score incorporates 15 clinical, electrocardiographic, and genetic variables. Recent validations demonstrate superior predictive accuracy (C-statistic > 0.80) for major arrhythmic events compared to older models [30].
- Machine Learning Models: Emerging tools like the BRUGADA-RISK calculator utilize machine learning algorithms to integrate dynamic ECG changes and clinical variables, offering personalized 5-year SCD risk estimates [31].
Evidence-Based Management and SCD Prevention
Implantable Cardioverter-Defibrillators (ICDs)
The implantable cardioverter-defibrillator remains the only therapy proven to prevent SCD in high-risk BrS patients. According to the 2022 ESC and 2017 AHA/ACC/HRS guidelines, ICD implantation is recommended as follows [2, 32]:
- Class I: Survivors of aborted SCA or patients with documented spontaneous sustained VT/VF.
- Class IIa: Patients with a spontaneous Type 1 ECG and a history of arrhythmic syncope.
- Class IIb: Asymptomatic patients with a spontaneous Type 1 ECG who have inducible VF during EPS.
Because BrS patients are typically young and lack structural heart disease, they are ideal candidates for the subcutaneous ICD (S-ICD). The S-ICD avoids the long-term risks of transvenous lead failure and systemic infection, which are significant in a population with a life expectancy of several decades post-implantation [33]. However, careful pre-implantation ECG screening is mandatory to ensure appropriate sensing and avoid T-wave oversensing, which is more common in BrS due to dynamic repolarization abnormalities.
Pharmacological Therapy: Quinidine and Isoproterenol
While ICDs terminate life-threatening arrhythmias, they do not prevent them. Pharmacotherapy plays a crucial role in reducing arrhythmic burden and managing electrical storms.
Quinidine: As a potent blocker of the transient outward potassium current ($I_{to}$), quinidine directly counteracts the fundamental electrophysiological derangement in BrS. It is highly effective in suppressing spontaneous VF and normalizing the ECG [34]. Quinidine is indicated (Class IIa) for patients with recurrent ICD shocks, those with a contraindication to ICD implantation, or as a bridge to ablation. Its use is limited primarily by gastrointestinal intolerance (diarrhea) and the risk of proarrhythmia (QT prolongation), necessitating careful initiation [35].
Isoproterenol: In the acute setting of an electrical storm, a continuous intravenous infusion of isoproterenol (1–2 mcg/min) is the first-line therapy. By stimulating beta-adrenergic receptors, isoproterenol increases the L-type calcium current ($I_{Ca,L}$) and heart rate, effectively counteracting the unopposed $I_{to}$, restoring the epicardial action potential dome, and suppressing phase 2 reentry [36].
Epicardial Substrate Ablation
One of the most significant advancements in BrS management is the development of epicardial RVOT substrate ablation. Pioneered by Nademanee and colleagues, this approach targets the delayed, fractionated electrograms in the RVOT epicardium [13]. Using high-density electroanatomical mapping, the arrhythmogenic substrate is identified and ablated using radiofrequency energy.
Long-term registry data demonstrate that epicardial ablation normalizes the ECG phenotype in >80% of patients and drastically reduces the incidence of recurrent VF [37]. The BRAVE (Brugada Ablation of VF Episodes) trial and other prospective studies have solidified substrate ablation as a Class IIb recommendation for patients with recurrent ICD shocks refractory to quinidine, and increasingly as an earlier intervention in high-risk cohorts [38].
Special Populations and Future Directions
The Asymptomatic Patient
The management of asymptomatic patients with a spontaneous Type 1 ECG remains the most vexing clinical dilemma. The annual risk of SCD is low (~0.8%), yet the cumulative lifetime risk is non-trivial. Conversely, the complication rate of ICDs in young, active patients (inappropriate shocks, lead fractures, infections) can exceed 10% over a decade [39]. Shared decision-making is paramount. For intermediate-risk patients who do not meet strict ICD criteria, the use of Implantable Loop Recorders (ILRs) is increasingly recommended (Class IIa/IIb) to monitor for asymptomatic arrhythmias and guide future interventions [2].
Pediatric and Female Populations
BrS exhibits a profound male predominance (up to 8:1), largely attributed to the influence of testosterone, which enhances $I_{to}$ expression [40]. Women with BrS generally have a lower penetrance and a more benign clinical course, though they are more likely to present with conduction system disease (e.g., sinus node dysfunction) rather than VF [41].
In pediatric populations, the presentation is distinct. Fever is the most critical trigger for arrhythmias in children with BrS. Elevated temperatures accelerate the kinetics of the mutated sodium channel, exacerbating the LOF phenotype. Immediate administration of antipyretics and continuous ECG monitoring during febrile illnesses are mandatory Class I recommendations for all BrS patients, but are especially critical in pediatrics [42].
Future Directions: Mapping and Gene Therapy
The future of BrS management lies in precision medicine. Non-invasive Electrocardiographic Imaging (ECGI) is emerging as a tool to map the epicardial RVOT substrate without the need for invasive epicardial puncture, potentially guiding prophylactic ablation [43]. Furthermore, as the genetic architecture of BrS becomes clearer, allele-specific gene silencing and CRISPR/Cas9-mediated gene editing of SCN5A mutations represent the ultimate frontier, offering the promise of a definitive cure rather than mere palliation of symptoms [44].
References
- Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20(6):1391-1396.
- Zeppenfeld K, Tfelt-Hansen J, de Riva M, et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2022;43(40):3997-4126.
- Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference. Circulation. 2005;111(5):659-670.
- Vatta M, Dumaine R, Varghese G, et al. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet. 2002;11(3):337-345.
- Probst V, Veltmann C, Eckardt L, et al. Long-term prognosis of patients diagnosed with Brugada syndrome: Results from the FINGER Brugada Syndrome Registry. Circulation. 2010;121(5):635-643.
- Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392(6673):293-296.
- Cerrone M, Remme CA, Tadros R, et al. Beyond the One Gene-One Disease Paradigm: Complex Genetics and Pleiotropy in Inheritable Cardiac Disorders. J Am Coll Cardiol. 2022;80(16):1564-1576.
- Hosseini SM, Kim R, Udupa S, et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death: Evidence-Based Evaluation of Gene Validity for Brugada Syndrome. Circulation. 2018;138(12):1195-1205.
- Barc J, Tadros R, Glinge C, et al. Genome-wide association analyses identify new Brugada syndrome risk loci and highlight a new mechanism of sodium channel regulation in disease susceptibility. Nat Genet. 2022;54(3):232-239.
- Kukavica D, Sommariva E, et al. Polygenic risk scores in Brugada syndrome: clinical implications and future perspectives. Europace. 2024;26(2):euad385.
- Antzelevitch C. Brugada syndrome. Pacing Clin Electrophysiol. 2006;29(10):1130-1159.
- Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100(15):1660-1666.
- Nademanee K, Veerakul G, Chandanamattha P, et al. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation. 2011;123(12):1270-1279.
- Coronel R, Casini S, Koopmann TT, et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, immunohistochemical, and computational study. Circulation. 2005;112(18):2769-2777.
- Sommariva E, Pappone C, Brugada J, et al. Microstructural abnormalities in Brugada syndrome: the overlap with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2021;42(34):3413-3423.
- Behr ER, Savio-Galimberti E, et al. Structural overlap between Brugada syndrome and arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2021;42(34):3424-3426.
- Gaita F, Giustetto C, Bianchi F, et al. Short QT Syndrome: a familial cause of sudden death. Circulation. 2003;108(8):965-970. [Note: High lead utility validated in subsequent cohorts].
- Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm. 2013;10(12):1932-1963.
- Kawada S, Morita H, Antzelevitch C, et al. Shanghai Score System for diagnosis of Brugada syndrome: Validation of the score system and system and reclassification of the patients. Heart Rhythm. 2018;15(8):1180-1187.
- Conte G, Sieira J, Ciconte G, et al. Drug-induced Brugada syndrome children: clinical features, device-based management, and long-term follow-up. J Am Coll Cardiol. 2014;63(21):2272-2279.
- Viskin S,