A novel potentially life-threatening cardiac arrhythmia syndrome identified with ECG
The ECG may be used to diagnose several cardiac genetic disorders, such as Brugada syndrome, long QT syndrome [LQTS] and early repolarization syndrome. These conditions have an underlying genetic basis and may precipitate lethal arrhythmias. Long QT syndrome was identified several decades ago by Jervell et al. Brugada syndrome was identified in the early 1990s and early repolarization was identified as a risk factor for sudden cardiac arrest in 2008 (1, 2, 3). In November 2018 researchers from Denmark, Netherlands and United Kingdom reported a novel ECG syndrome characterized by widespread ST-segment depressions and an increased risk of sudden cardiac arrest. They identified five unrelated families with features that represent a previously unrecognized autosomal dominant syndrome (4).
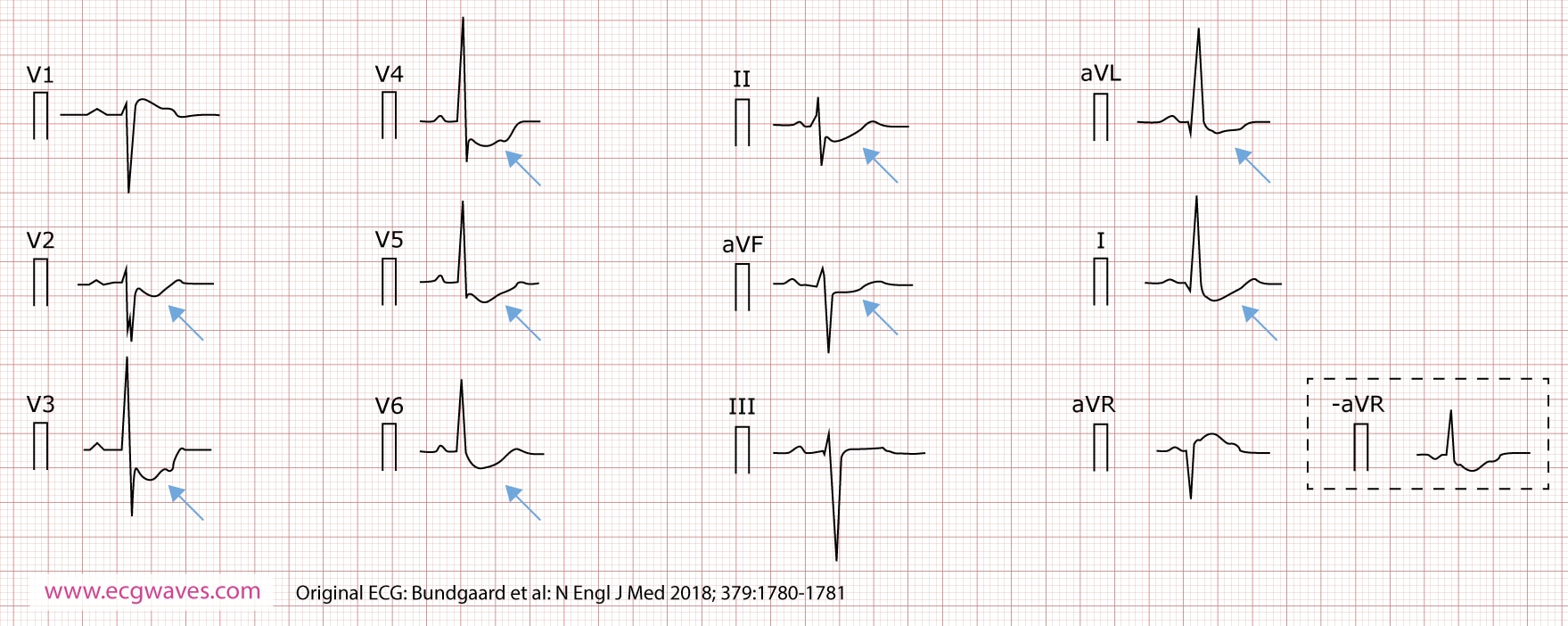
A male patient (Figure 1) presented at age 36 years with an ECG showing deep and persistent, concave-upward ST-segment depression in leads I, II, aVL, aVF, and V2 through V6. These ECG changes were stable over a period of 30 years. The ST-segment depressions were accentuated during exercise. The patient developed atrial fibrillation at 55 years of age. At age 64 he developed ventricular fibrillation and aborted sudden cardiac death. His son and daughter presented with similar ECG changes. Four additional family members presented with similar ECG changes. Two relatives died from sudden cardiac death. Atrial fibrillation and ventricular arrhythmias were observed in the fifth decade of life in several family members. Imaging studies did not reveal structural cardiac abnormalities, nor coronary artery disease.
Another male patient, with similar ST-segment changes, experienced syncopal episodes, ventricular tachycardia (including torsades des pointes), ventricular-fibrillation arrests and moderate left ventricular dysfunction. This patient died at age 17 years due to complications related to his arrhythmia. His father and uncle, with similar ST-segment changes, were diagnosed with atrial fibrillation in their 60s. The father had repeated episodes of ventricular tachycardia and ventricular fibrillation. Several relatives presented with identical ECG changes (the youngest at age 6 years).
Another 9 affected individuals were presented in the study. All genetic tests, using gene panels for known gene mutations for cardiovascular disease, were negative.
Summary
- Bundgaard et al have presumably identified a novel familial (autosomal dominant) cardiac syndrome. The underlying genetic abnormality is yet to be discovered.
- Diagnosis of this syndrome is based on ECG, family history and clinical presentation.
- Patients present with syncope, early onset of atrial fibrillation, ventricular arrhythmias and sudden cardiac arrest.
- Given the prevalence of atrial arrhythmias, ventricular arrhythmias and sudden cardiac arrests, this syndrome appears to be very serious.
- The ECG is characterized by marked, persistent, nonischemic ST-segment depression. These ST-segment changes are stable over time (as compared with Brugada syndrome and LQTS, which both exhibit highly dynamic ECG changes).
Related topics
Ischemic and nonischemic ST-segment depressions
Author
Dr Araz Rawshani, MD, PhD
University of Gothenburg, The Sahlgrenska University Hospital, Sweden
References
- Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J 1957;54:59-68.
- Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome — a multicenter report. J Am Coll Cardiol 1992;20:1391-1396.
- Haïssaguerre M, Derval N, Sacher F, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med 2008;358:2016-2023.
- Bundgaard et al. A Novel Familial Cardiac Arrhythmia Syndrome with Widespread ST-Segment Depression. N Engl J Med 2018; 379:1780-1781.